

Identifying differentially expressed transcripts from RNA-seq data with biological variation
- PMID: 22563066
- PMCID: PMC3381971
- DOI: 10.1093/bioinformatics/bts260
Identifying differentially expressed transcripts from RNA-seq data with biological variation
Abstract
Motivation: High-throughput sequencing enables expression analysis at the level of individual transcripts. The analysis of transcriptome expression levels and differential expression (DE) estimation requires a probabilistic approach to properly account for ambiguity caused by shared exons and finite read sampling as well as the intrinsic biological variance of transcript expression.
Results: We present Bayesian inference of transcripts from sequencing data (BitSeq), a Bayesian approach for estimation of transcript expression level from RNA-seq experiments. Inferred relative expression is represented by Markov chain Monte Carlo samples from the posterior probability distribution of a generative model of the read data. We propose a novel method for DE analysis across replicates which propagates uncertainty from the sample-level model while modelling biological variance using an expression-level-dependent prior. We demonstrate the advantages of our method using simulated data as well as an RNA-seq dataset with technical and biological replication for both studied conditions.
Availability: The implementation of the transcriptome expression estimation and differential expression analysis, BitSeq, has been written in C++ and Python. The software is available online from http://code.google.com/p/bitseq/, version 0.4 was used for generating results presented in this article.
Figures
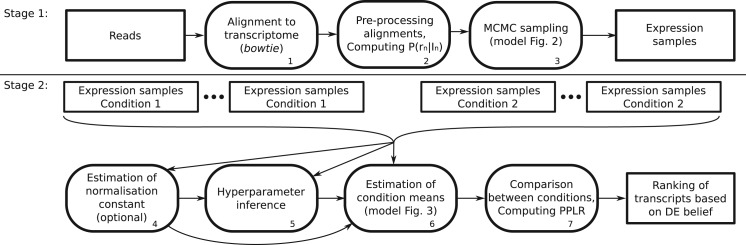
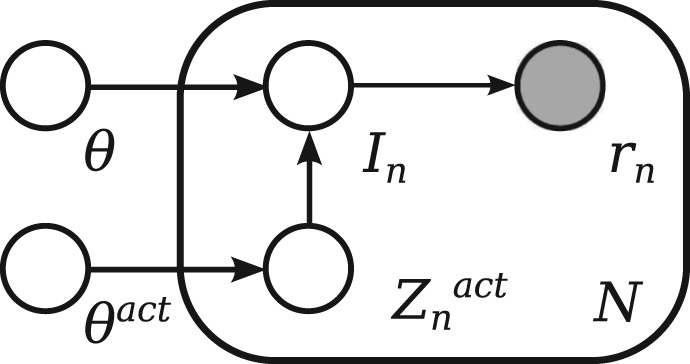
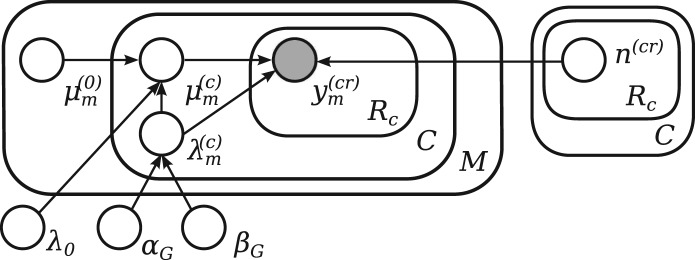
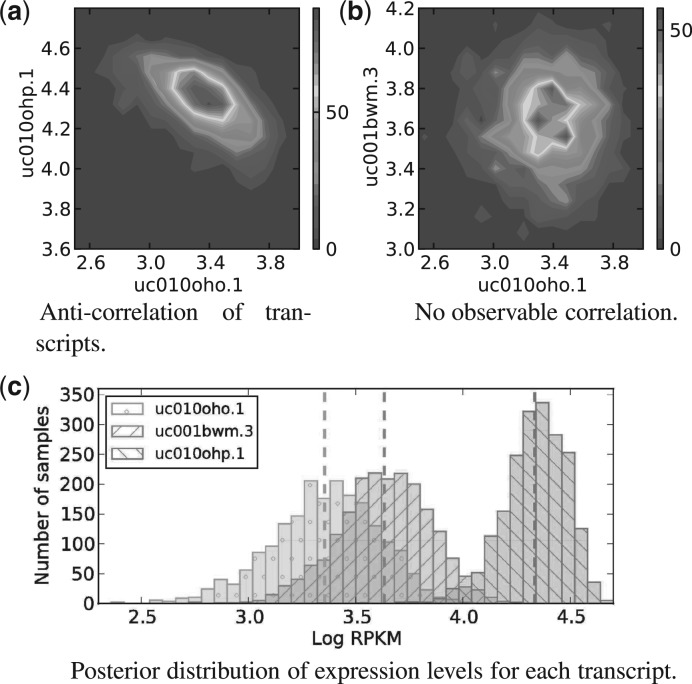
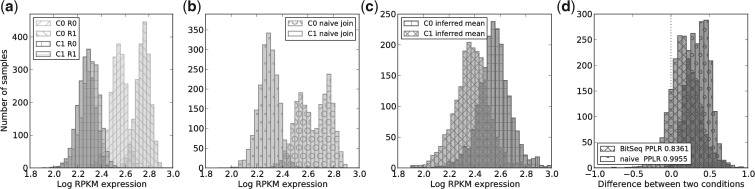
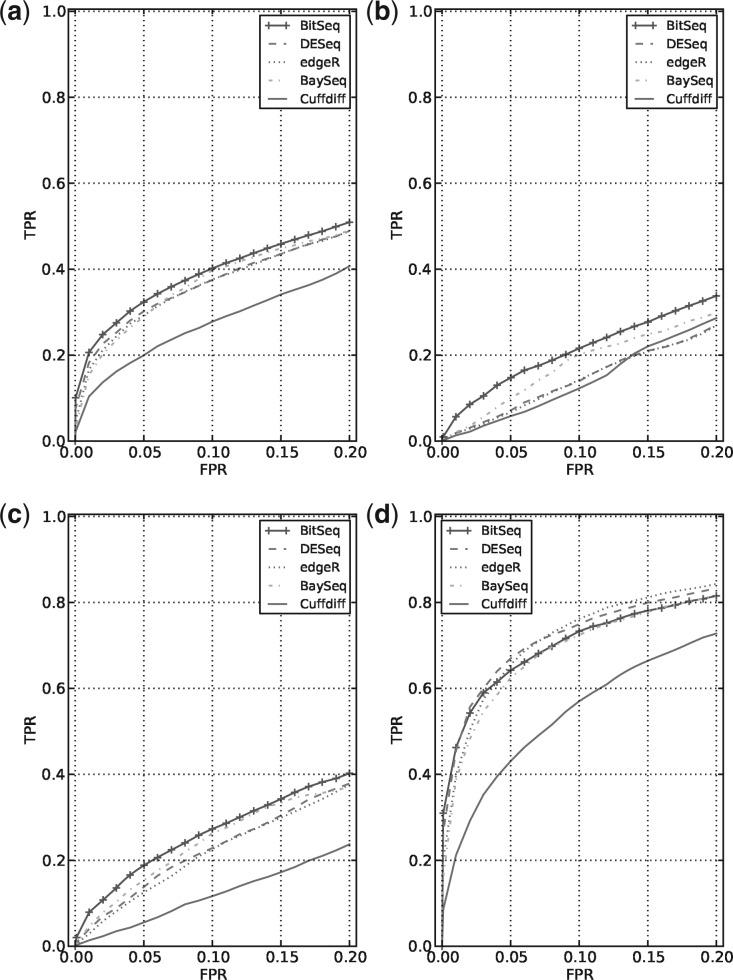
References
-
- Cleveland W. S. LOWESS: a program for smoothing scatterplots by robust locally weighted regression. Am. Stat. 1981;35:54.
-
- Cloonan N., et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods. 2008;5:613–619. - PubMed
-
- Gelman A., et al. Bayesian Data Analysis. 2. Chapman and Hall/CRC; 2003.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
