

Alternative splicing of Spg7, a gene involved in hereditary spastic paraplegia, encodes a variant of paraplegin targeted to the endoplasmic reticulum
- PMID: 22563492
- PMCID: PMC3341365
- DOI: 10.1371/journal.pone.0036337
Alternative splicing of Spg7, a gene involved in hereditary spastic paraplegia, encodes a variant of paraplegin targeted to the endoplasmic reticulum
Abstract
Background: Hereditary spastic paraplegia defines a group of genetically heterogeneous diseases characterized by weakness and spasticity of the lower limbs owing to retrograde degeneration of corticospinal axons. One autosomal recessive form of the disease is caused by mutation in the SPG7 gene. Paraplegin, the product of SPG7, is a component of the m-AAA protease, a high molecular weight complex that resides in the mitochondrial inner membrane, and performs crucial quality control and biogenesis functions in mitochondria.
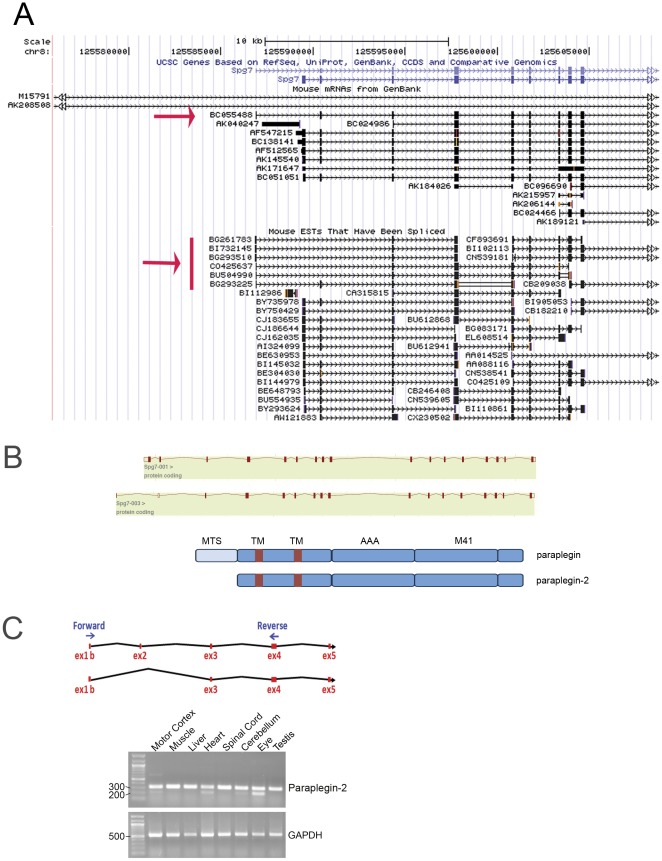
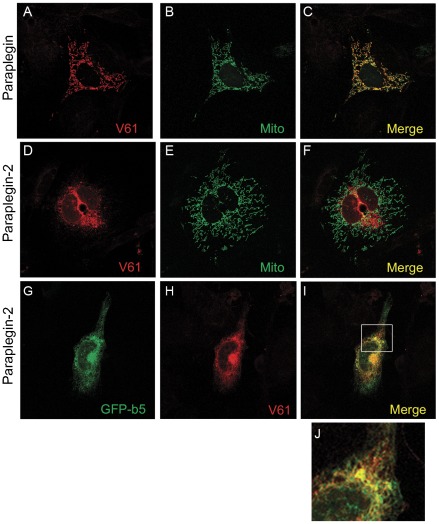
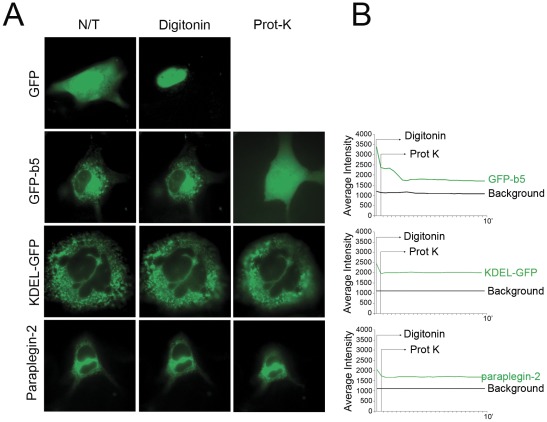
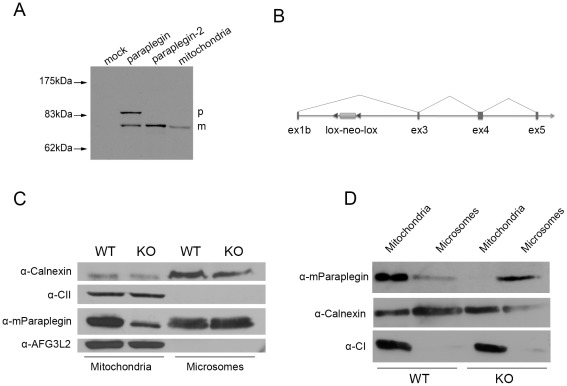
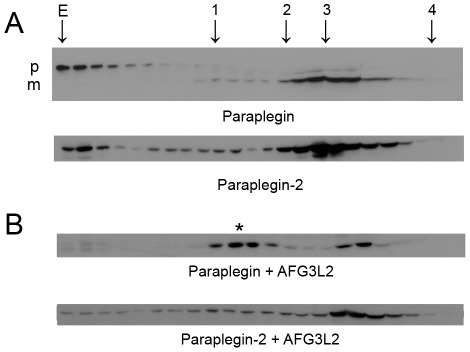
Principal findings: Here we show the existence in the mouse of a novel isoform of paraplegin, which we name paraplegin-2, encoded by alternative splicing of Spg7 through usage of an alternative first exon. Paraplegin-2 lacks the mitochondrial targeting sequence, and is identical to the mature mitochondrial protein. Remarkably, paraplegin-2 is targeted to the endoplasmic reticulum. We find that paraplegin-2 exposes the catalytic domains to the lumen of the endoplasmic reticulum. Moreover, endogenous paraplegin-2 accumulates in microsomal fractions prepared from mouse brain and retina. Finally, we show that the previously generated mouse model of Spg7-linked hereditary spastic paraplegia is an isoform-specific knock-out, in which mitochondrial paraplegin is specifically ablated, while expression of paraplegin-2 is retained.
Conclusions/significance: These data suggest a possible additional role of AAA proteases outside mitochondria and open the question of their implication in neurodegeneration.
Conflict of interest statement
Figures
References
-
- Harding AE. Hereditary spastic paraplegias. Semin Neurol. 1993;13:333–336. - PubMed
-
- Reid E, Rugarli EI. In: Valle, Beaudet, Vogelstein, Kinzler, Antonarakis, et al., editors. Scriver's Online Metabolic and Molecular Bases of Inherited Diseases; 2010.
-
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. - PubMed
-
- Arnoldi A, Tonelli A, Crippa F, Villani G, Pacelli C, et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum Mutat. 2008;29:522–531. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
