

A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism
- PMID: 22566635
- PMCID: PMC3361440
- DOI: 10.1073/pnas.1120210109
A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism
Abstract
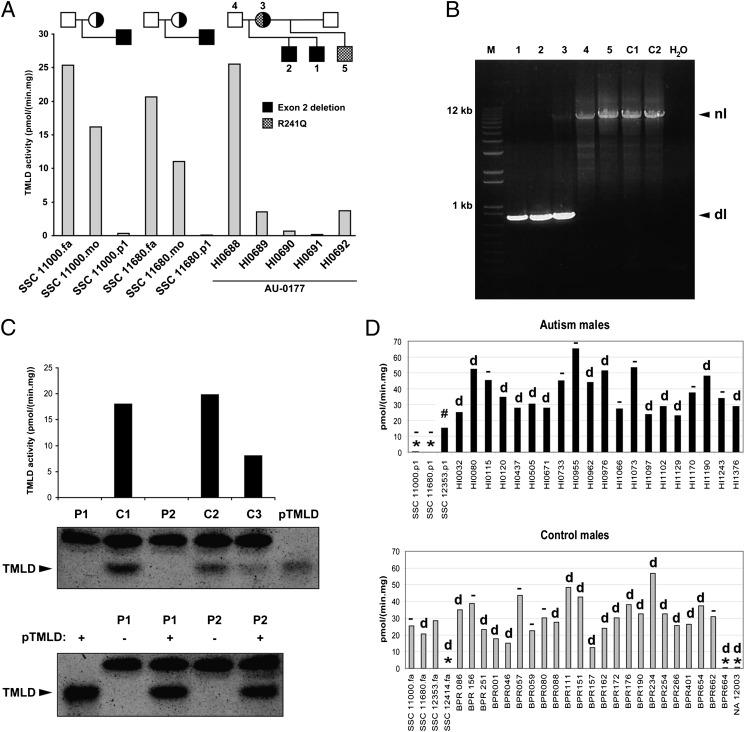
We recently reported a deletion of exon 2 of the trimethyllysine hydroxylase epsilon (TMLHE) gene in a proband with autism. TMLHE maps to the X chromosome and encodes the first enzyme in carnitine biosynthesis, 6-N-trimethyllysine dioxygenase. Deletion of exon 2 of TMLHE causes enzyme deficiency, resulting in increased substrate concentration (6-N-trimethyllysine) and decreased product levels (3-hydroxy-6-N-trimethyllysine and γ-butyrobetaine) in plasma and urine. TMLHE deficiency is common in control males (24 in 8,787 or 1 in 366) and was not significantly increased in frequency in probands from simplex autism families (9 in 2,904 or 1 in 323). However, it was 2.82-fold more frequent in probands from male-male multiplex autism families compared with controls (7 in 909 or 1 in 130; P = 0.023). Additionally, six of seven autistic male siblings of probands in male-male multiplex families had the deletion, suggesting that TMLHE deficiency is a risk factor for autism (metaanalysis Z-score = 2.90 and P = 0.0037), although with low penetrance (2-4%). These data suggest that dysregulation of carnitine metabolism may be important in nondysmorphic autism; that abnormalities of carnitine intake, loss, transport, or synthesis may be important in a larger fraction of nondysmorphic autism cases; and that the carnitine pathway may provide a novel target for therapy or prevention of autism.
Conflict of interest statement
Conflict of interest statement: Multiple authors are based in the Department of Molecular and Human Genetics at Baylor College of Medicine, which offers extensive genetic laboratory testing, and Baylor College of Medicine derives revenue from this activity. The department offers biochemical and molecular diagnostic testing for trimethyllysine hydroxylase, epsilon gene deficiency. P.B.S.C.-S., S.V., F.M.V., and A.L.B. have filed a patent related to some of the work reported.
Figures



References
-
- Conference Proceedings (2004) Carnitine. The science behind a conditionally essential nutrient. Proceedings of a conference. March 25–26, 2004. Bethesda, MD. Ann N Y Acad Sci 1033:1–197. - PubMed
-
- Slonim AE, et al. Dietary-dependent carnitine deficiency as a cause of nonketotic hypoglycemia in an infant. J Pediatr. 1981;99:551–555. - PubMed
-
- Schmidt-Sommerfeld E, Penn D. Carnitine and total parenteral nutrition of the neonate. Biol Neonate. 1990;58(Suppl 1):81–88. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
