

Chronic inflammation in cancer development
- PMID: 22566887
- PMCID: PMC3342348
- DOI: 10.3389/fimmu.2011.00098
Chronic inflammation in cancer development
Abstract
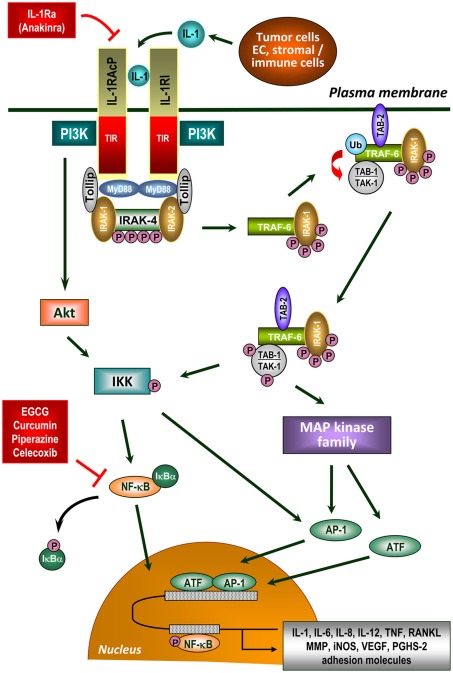
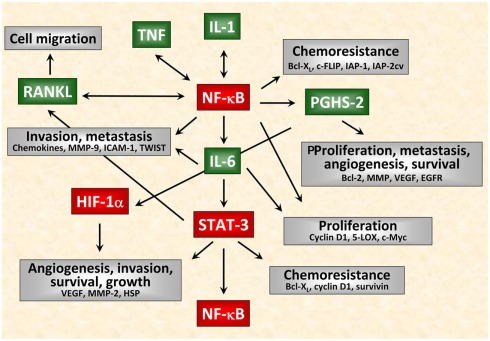
Chronic inflammatory mediators exert pleiotropic effects in the development of cancer. On the one hand, inflammation favors carcinogenesis, malignant transformation, tumor growth, invasion, and metastatic spread; on the other hand inflammation can stimulate immune effector mechanisms that might limit tumor growth. The link between cancer and inflammation depends on intrinsic and extrinsic pathways. Both pathways result in the activation of transcription factors such as NF-κB, STAT-3, and HIF-1 and in accumulation of tumorigenic factors in tumor and microenvironment. STAT-3 and NF-κB interact at multiple levels and thereby boost tumor-associated inflammation which can suppress anti-tumor immune responses. These factors also promote tumor growth, progression, and metastatic spread. IL-1, IL-6, TNF, and PGHS-2 are key mediators of an inflammatory milieu by modulating the expression of tumor-promoting factors. In this review we concentrate on the crucial role of pro-inflammatory mediators in inflammation-driven carcinogenesis and outline molecular mechanisms of IL-1 signaling in tumors. In addition, we elucidate the dual roles of stress proteins as danger signals in the development of anti-cancer immunity and anti-apoptotic functions.
Keywords: IL-1; NF-κB; STAT-3; carcinogenesis; heat shock proteins; inflammation; tumorigenic factors.
Figures
References
-
- Aaltoma S. H., Lipponen P. K., Kosma V. M. (2001). Inducible nitric oxide synthase (iNOS) expression and its prognostic value in prostate cancer. Anticancer Res. 21, 3101–3106 - PubMed
-
- Adhami V. M., Malik A., Zaman N., Sarfaraz S., Siddiqui I. A., Syed D. N., Afaq F., Pasha F. S., Saleem M., Mukhtar H. (2007). Combined inhibitory effects of green tea polyphenols and selective cyclooxygenase-2 inhibitors on the growth of human prostate cancer cells both in vitro and in vivo. Clin. Cancer Res. 13, 1611–161910.1158/1078-0432.CCR-06-2269 - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous