

PITX2 and FOXC1 spectrum of mutations in ocular syndromes
- PMID: 22569110
- PMCID: PMC3499749
- DOI: 10.1038/ejhg.2012.80
PITX2 and FOXC1 spectrum of mutations in ocular syndromes
Abstract
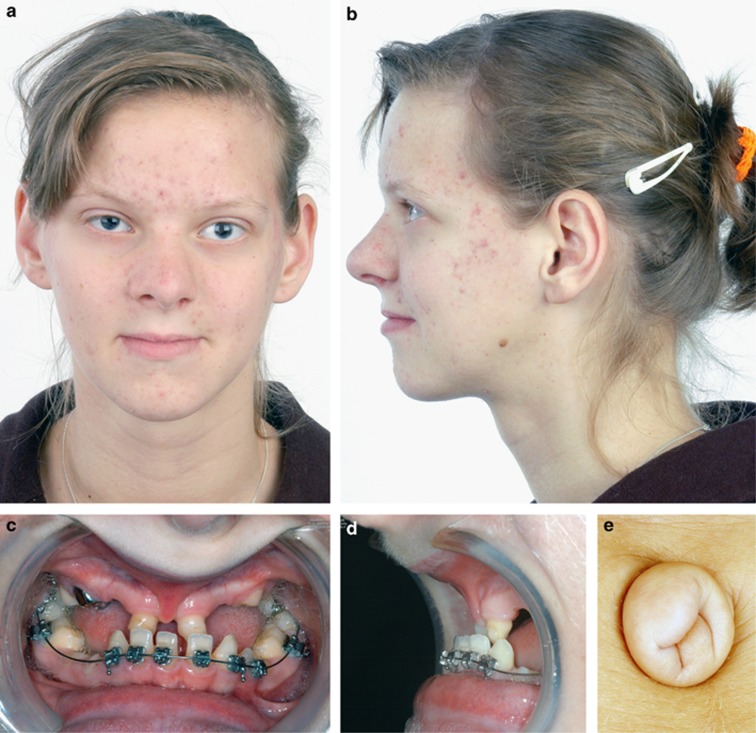
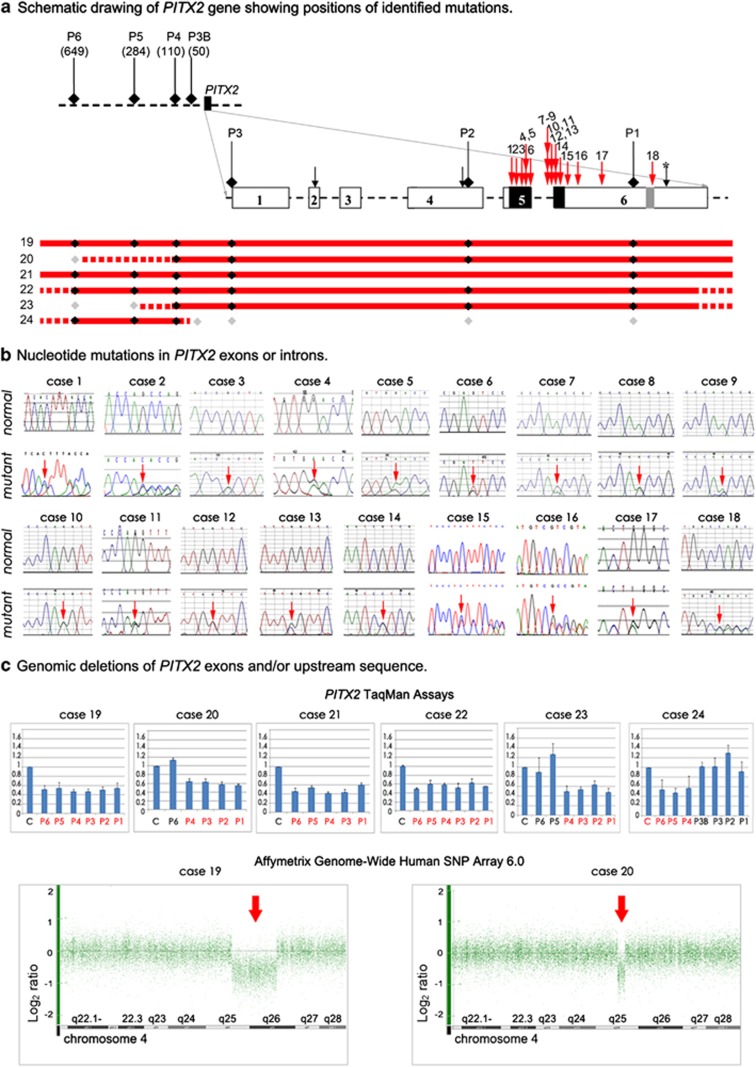
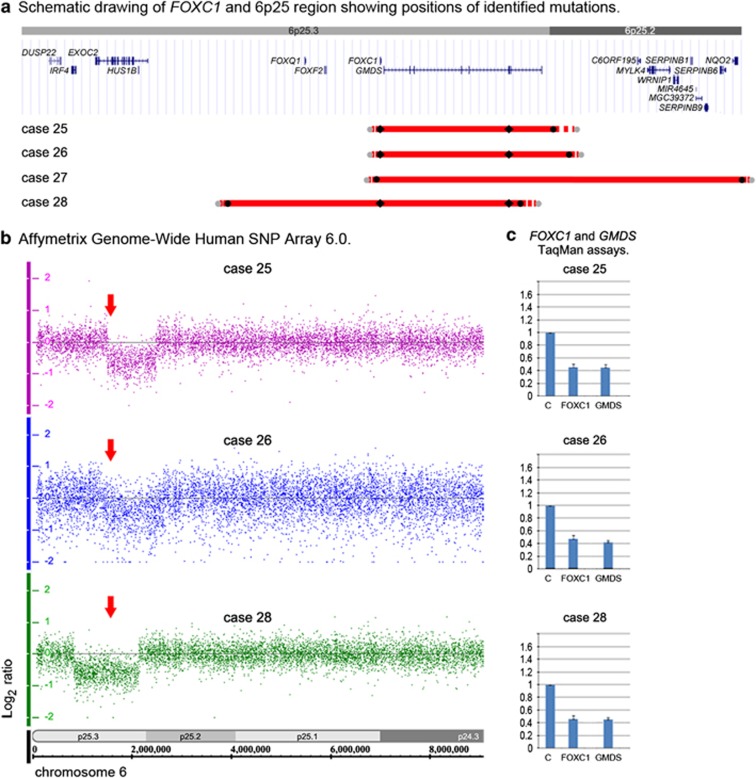
Anterior segment dysgenesis (ASD) encompasses a broad spectrum of developmental conditions affecting anterior ocular structures and associated with an increased risk for glaucoma. Various systemic anomalies are often observed in ASD conditions such as Axenfeld-Rieger syndrome (ARS) and De Hauwere syndrome. We report DNA sequencing and copy number analysis of PITX2 and FOXC1 in 76 patients with syndromic or isolated ASD and related conditions. PITX2 mutations and deletions were found in 24 patients with dental and/or umbilical anomalies seen in all. Seven PITX2-mutant alleles were novel including c.708_730del, the most C-terminal mutation reported to date. A second case of deletion of the distant upstream but not coding region of PITX2 was identified, highlighting the importance of this recently discovered mechanism for ARS. FOXC1 deletions were observed in four cases, three of which demonstrated hearing and/or heart defects, including a patient with De Hauwere syndrome; no nucleotide mutations in FOXC1 were identified. Review of the literature identified several other patients with 6p25 deletions and features of De Hauwere syndrome. The 1.3-Mb deletion of 6p25 presented here defines the critical region for this phenotype and includes the FOXC1, FOXF2, and FOXQ1 genes. In summary, PITX2 or FOXC1 disruptions explained 63% of ARS and 6% of other ASD in our cohort; all affected patients demonstrated additional systemic defects with PITX2 mutations showing a strong association with dental and/or umbilical anomalies and FOXC1 with heart and hearing defects. FOXC1 deletion was also found to be associated with De Hauwere syndrome.
Figures
References
-
- Sowden JC: Molecular and developmental mechanisms of anterior segment dysgenesis. Eye (Lond) 2007; 21: 1310–1318. - PubMed
-
- Rieger H: Verlagerung und schlitzform der pupille mit hypoplasie des irisvorderblattes. Z Augenheilkd 1934; 84: 98–103.
-
- Alward WL: Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol 2000; 130: 107–115. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
