

Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation
- PMID: 22570497
- PMCID: PMC3384190
- DOI: 10.1073/pnas.1202916109
Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation
Abstract
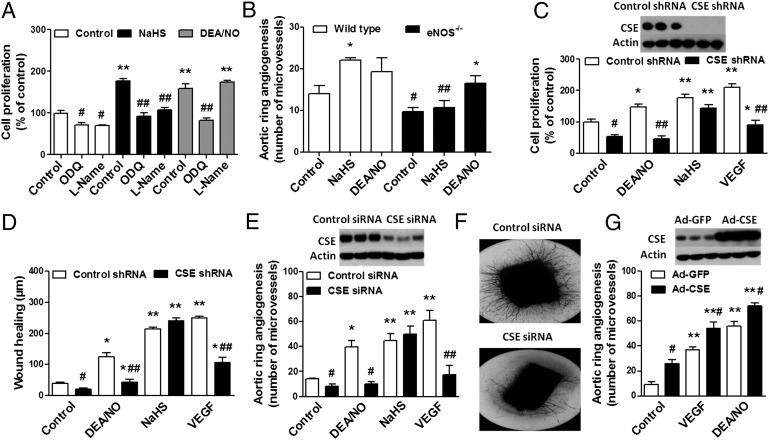
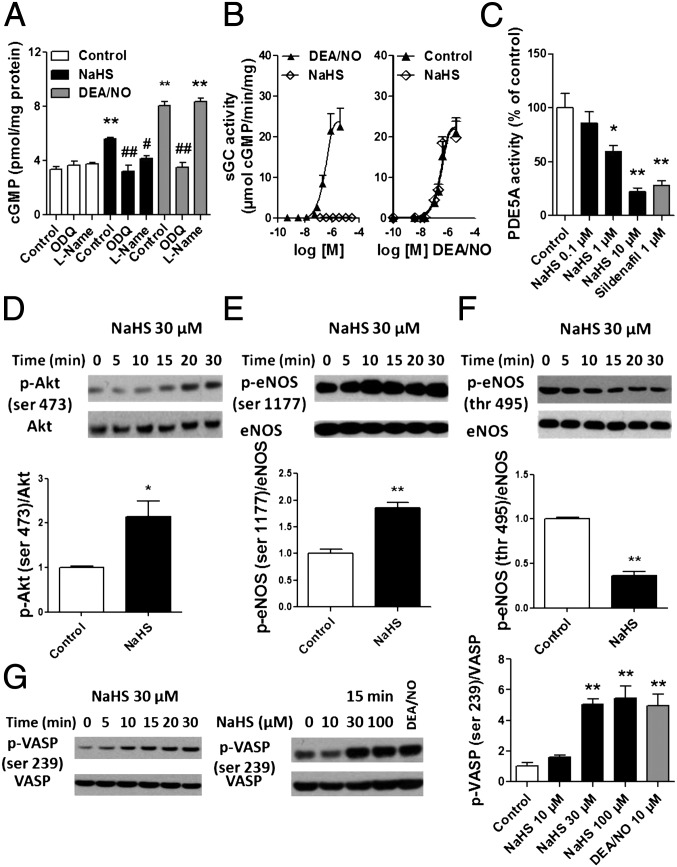
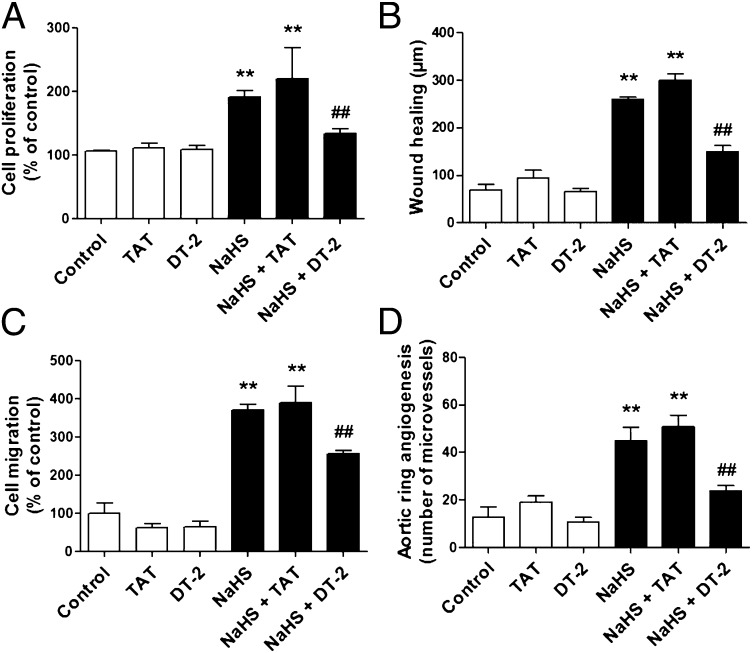
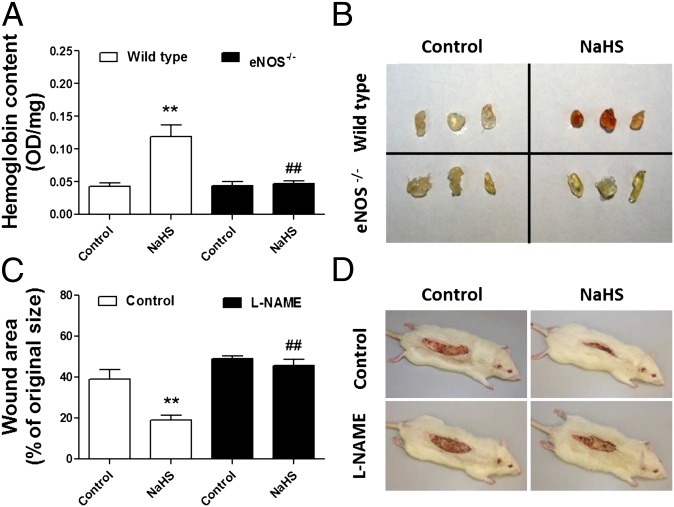
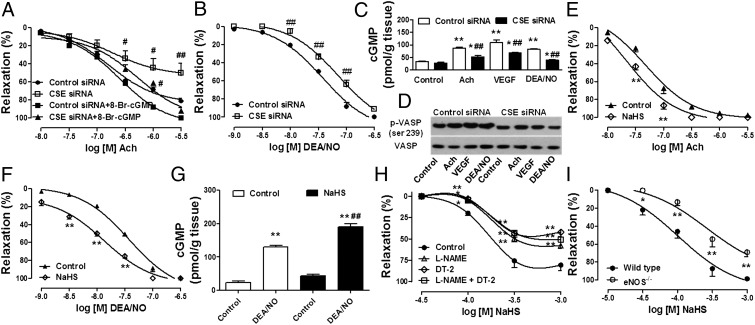
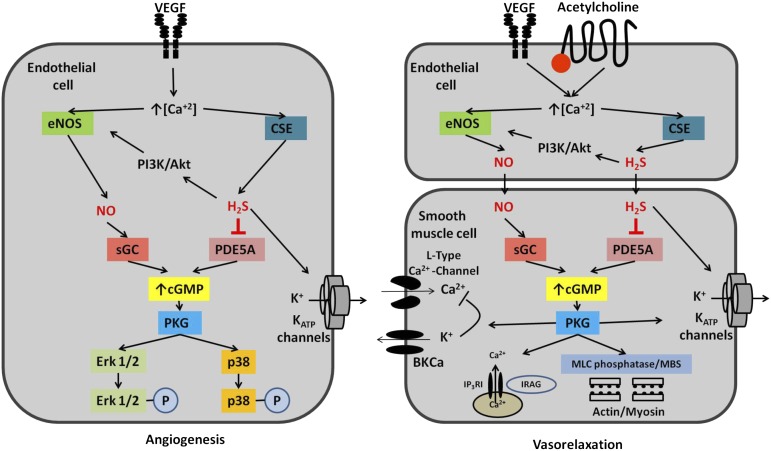
Hydrogen sulfide (H(2)S) is a unique gasotransmitter, with regulatory roles in the cardiovascular, nervous, and immune systems. Some of the vascular actions of H(2)S (stimulation of angiogenesis, relaxation of vascular smooth muscle) resemble those of nitric oxide (NO). Although it was generally assumed that H(2)S and NO exert their effects via separate pathways, the results of the current study show that H(2)S and NO are mutually required to elicit angiogenesis and vasodilatation. Exposure of endothelial cells to H(2)S increases intracellular cyclic guanosine 5'-monophosphate (cGMP) in a NO-dependent manner, and activated protein kinase G (PKG) and its downstream effector, the vasodilator-stimulated phosphoprotein (VASP). Inhibition of endothelial isoform of NO synthase (eNOS) or PKG-I abolishes the H(2)S-stimulated angiogenic response, and attenuated H(2)S-stimulated vasorelaxation, demonstrating the requirement of NO in vascular H(2)S signaling. Conversely, silencing of the H(2)S-producing enzyme cystathionine-γ-lyase abolishes NO-stimulated cGMP accumulation and angiogenesis and attenuates the acetylcholine-induced vasorelaxation, indicating a partial requirement of H(2)S in the vascular activity of NO. The actions of H(2)S and NO converge at cGMP; though H(2)S does not directly activate soluble guanylyl cyclase, it maintains a tonic inhibitory effect on PDE5, thereby delaying the degradation of cGMP. H(2)S also activates PI3K/Akt, and increases eNOS phosphorylation at its activating site S1177. The cooperative action of the two gasotransmitters on increasing and maintaining intracellular cGMP is essential for PKG activation and angiogenesis and vasorelaxation. H(2)S-induced wound healing and microvessel growth in matrigel plugs is suppressed by pharmacological inhibition or genetic ablation of eNOS. Thus, NO and H(2)S are mutually required for the physiological control of vascular function.
Conflict of interest statement
Conflict of interest statement: C.S. is a holder of patents related to the therapeutic effects of H2S.
Figures
Comment in
-
Shared signaling pathways among gasotransmitters.Proc Natl Acad Sci U S A. 2012 Jun 5;109(23):8801-2. doi: 10.1073/pnas.1206646109. Epub 2012 May 21. Proc Natl Acad Sci U S A. 2012. PMID: 22615409 Free PMC article. No abstract available.
References
-
- Ignarro LJ. Nitric oxide: A unique endogenous signaling molecule in vascular biology. Biosci Rep. 1999;19:51–71. - PubMed
-
- Bryan NS, Bian K, Murad F. Discovery of the nitric oxide signaling pathway and targets for drug development. Front Biosci. 2009;14:1–18. - PubMed
-
- Wang R. The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal. 2003;5:493–501. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
