

Osteogenesis Imperfecta: A Review with Clinical Examples
- PMID: 22570641
- PMCID: PMC3343766
- DOI: 10.1159/000332228
Osteogenesis Imperfecta: A Review with Clinical Examples
Abstract
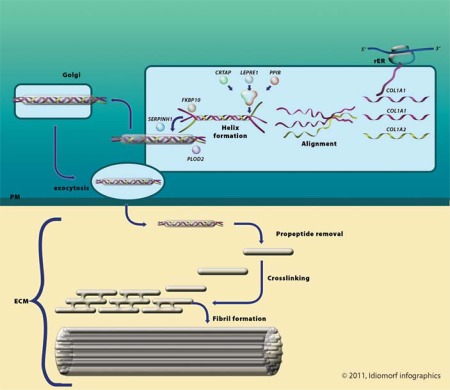
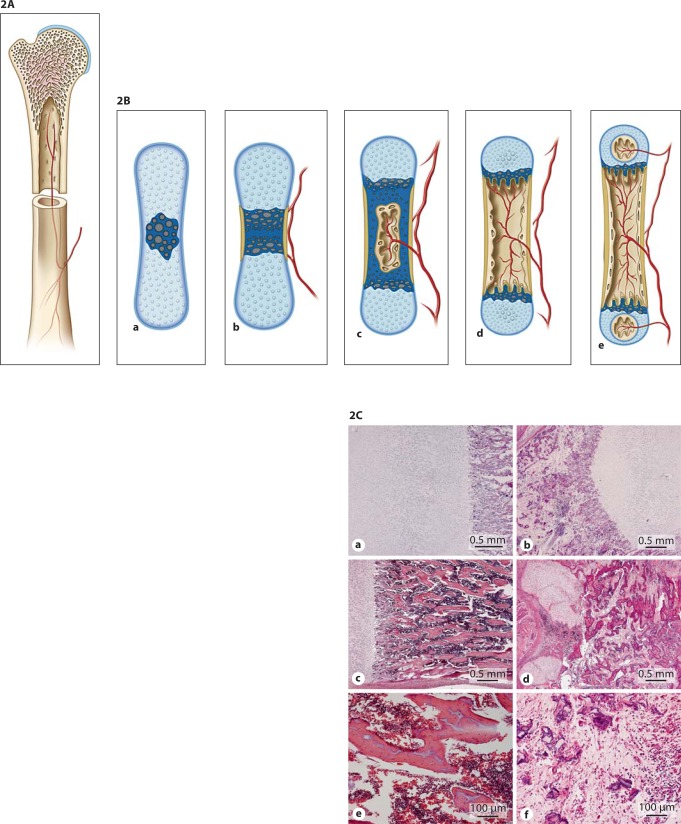
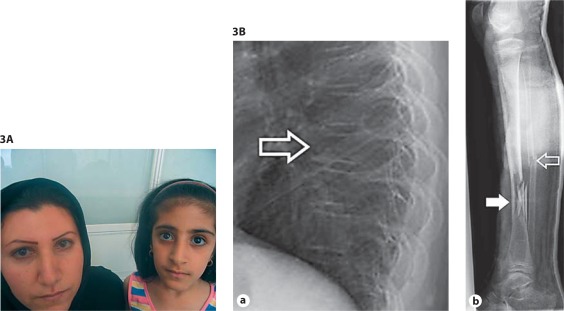
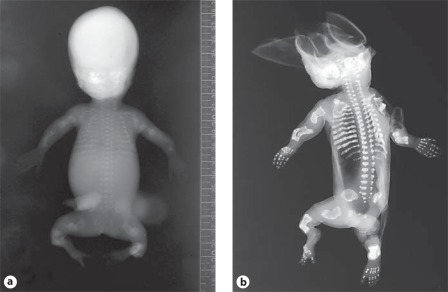
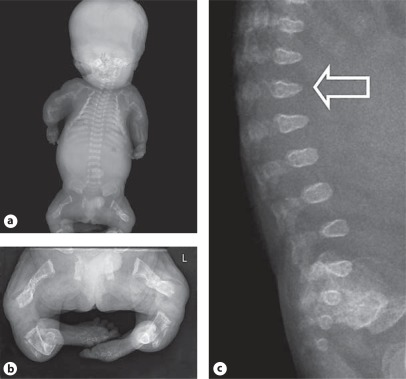
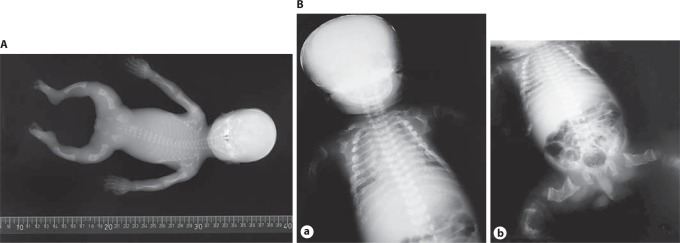
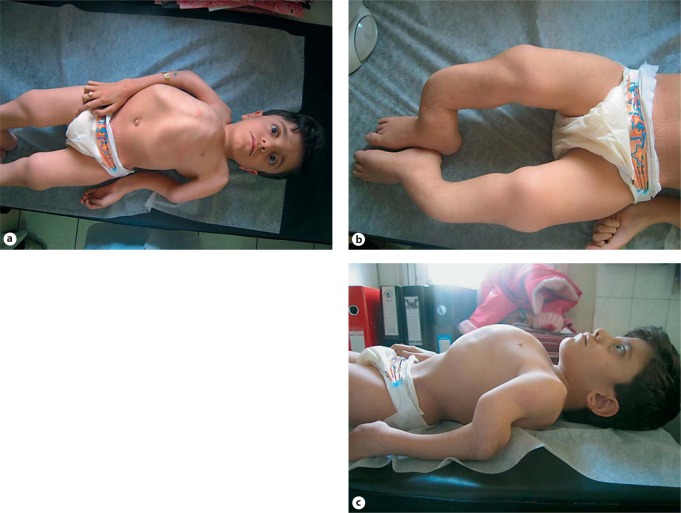
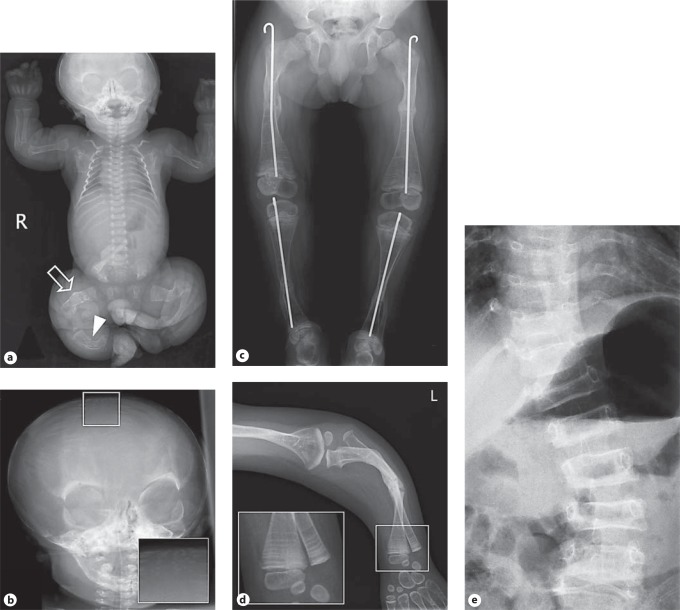
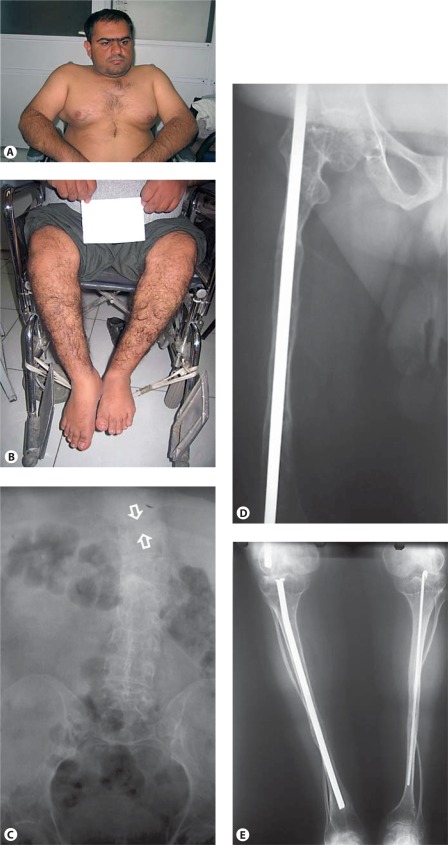
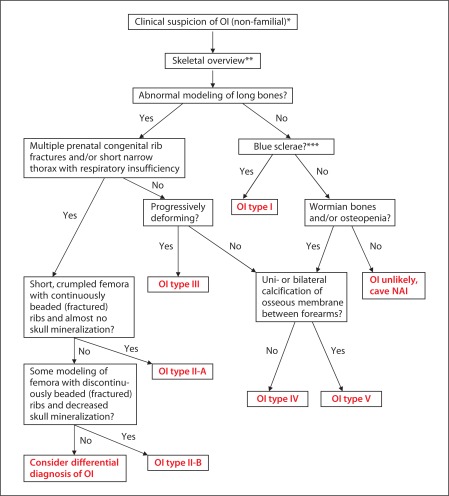
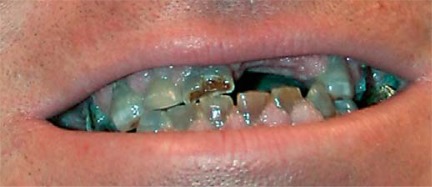
Osteogenesis imperfecta (OI) is characterized by susceptibility to bone fractures, with a severity ranging from subtle increase in fracture frequency to prenatal fractures. The first scientific description of OI dates from 1788. Since then, important milestones in OI research and treatment have, among others, been the classification of OI into 4 types (the 'Sillence classification'), the discovery of defects in collagen type I biosynthesis as a cause of most cases of OI and the use of bisphosphonate therapy. Furthermore, in the past 5 years, it has become clear that OI comprises a group of heterogeneous disorders, with an estimated 90% of cases due to a causative variant in the COL1A1 or COL1A2 genes and with the remaining 10% due to causative recessive variants in the 8 genes known so far, or in other currently unknown genes. This review aims to highlight the current knowledge around the history, epidemiology, pathogenesis, clinical/radiological features, management, and future prospects of OI. The text will be illustrated with clinical descriptions, including radiographs and, where possible, photographs of patients with OI.
Figures
References
-
- Aase JM. Diagnostic Dysmorphology. New York: Plenum Medical Book Company; 1990.
-
- Ablin DS, Greenspan A, Reinhart M, Grix A. Differentiation of child abuse from osteogenesis imperfecta. AJR Am J Roentgenol. 1990;154:1035–1046. - PubMed
-
- Baljet B. Willem Vrolik as a teratologist [in Dutch] Ned Tijdschr Geneeskd. 1984;128:1530–1534. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
