

SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon
- PMID: 22572391
- PMCID: PMC7102726
- DOI: 10.1016/j.coviro.2012.04.004
SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon
Abstract
SARS-CoV is a pathogenic coronavirus that emerged from a zoonotic reservoir, leading to global dissemination of the virus. The association SARS-CoV with aberrant cytokine, chemokine, and Interferon Stimulated Gene (ISG) responses in patients provided evidence that SARS-CoV pathogenesis is at least partially controlled by innate immune signaling. Utilizing models for SARS-CoV infection, key components of innate immune signaling pathways have been identified as protective factors against SARS-CoV disease, including STAT1 and MyD88. Gene transcription signatures unique to SARS-CoV disease states have been identified, but host factors that regulate exacerbated disease phenotypes still remain largely undetermined. SARS-CoV encodes several proteins that modulate innate immune signaling through the antagonism of the induction of Interferon and by avoidance of ISG effector functions.
Copyright © 2012. Published by Elsevier B.V.
Figures
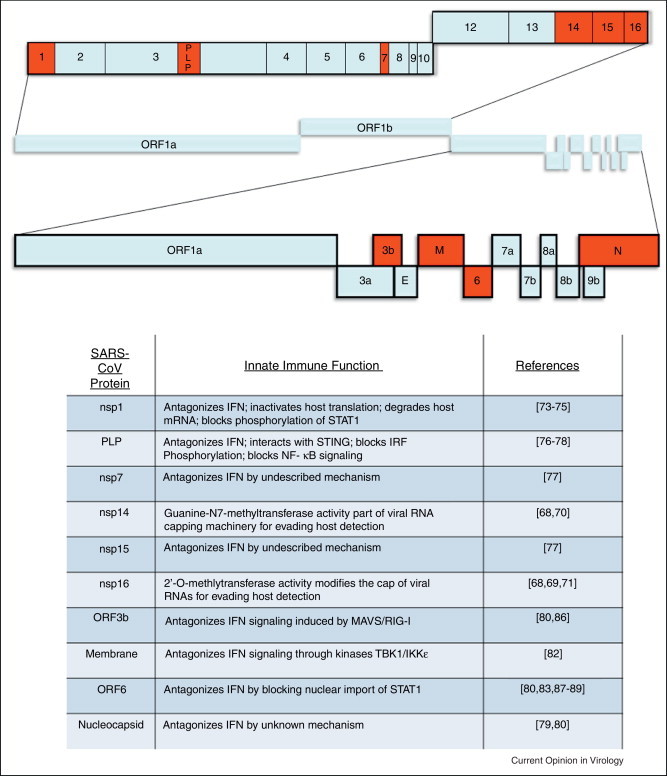
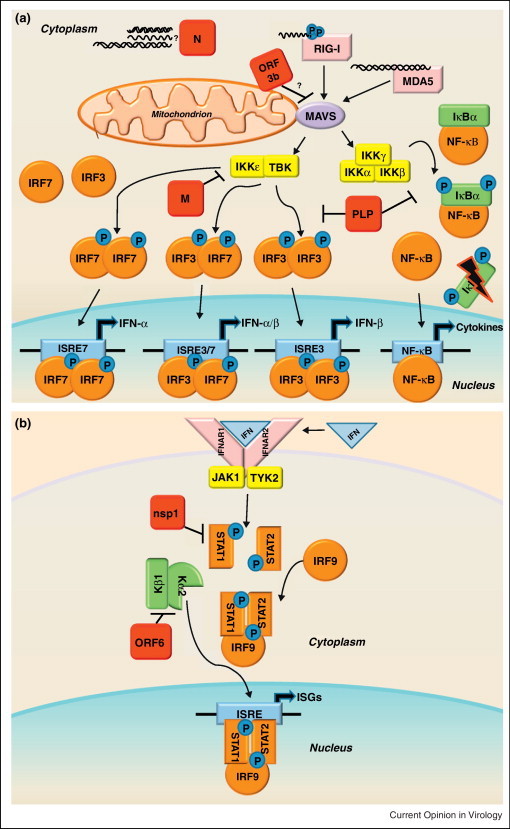
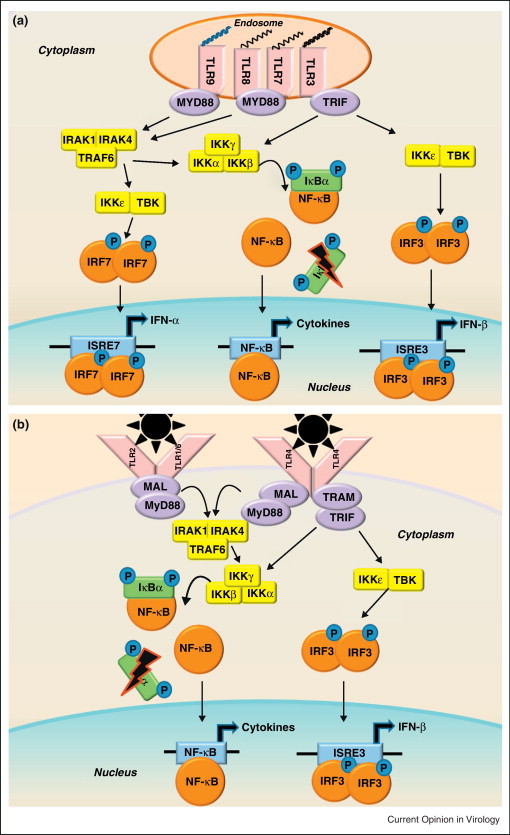
References
-
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.-R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A.M. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New England Journal of Medicine. 2003;348:1967–1976. - PubMed
-
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W. A novel coronavirus associated with severe acute respiratory syndrome. New England Journal of Medicine. 2003;348:1953–1966. - PubMed
-
- World Health Organization: Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003 on World Wide Web URL: http://www.who.int/csr/sars/country/table2004_04_21/en/index.html.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
