

Survivin is a therapeutic target in Merkel cell carcinoma
- PMID: 22572880
- PMCID: PMC3726222
- DOI: 10.1126/scitranslmed.3003713
Survivin is a therapeutic target in Merkel cell carcinoma
Abstract
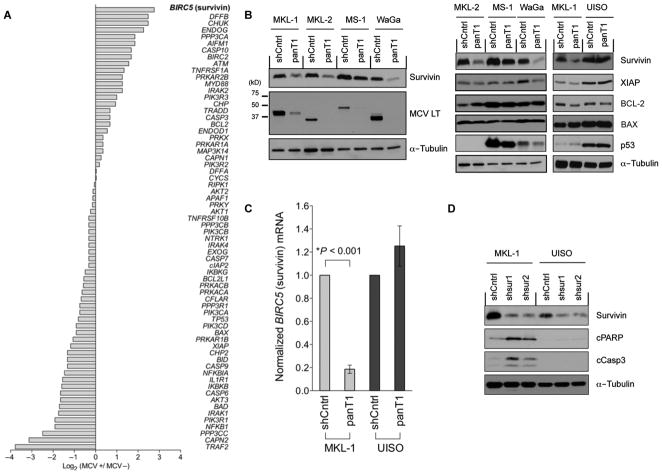
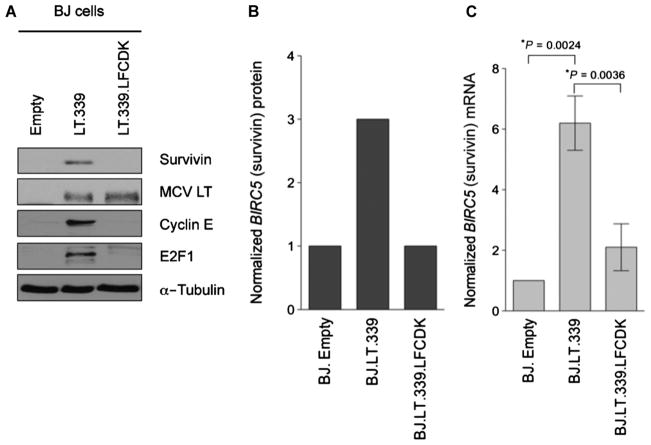
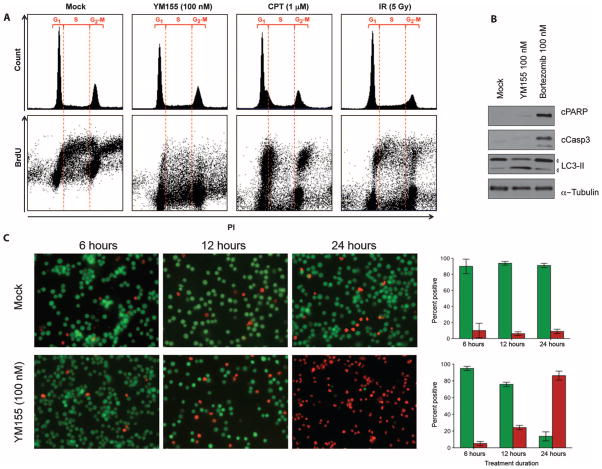
Merkel cell polyomavirus (MCV) causes ~80% of primary and metastatic Merkel cell carcinomas (MCCs). By comparing digital transcriptome subtraction deep-sequencing profiles, we found that transcripts of the cellular survivin oncoprotein [BIRC5a (baculoviral inhibitor of apoptosis repeat-containing 5)] were up-regulated sevenfold in virus-positive compared to virus-negative MCC tumors. Knockdown of MCV large T antigen in MCV-positive MCC cell lines decreased survivin mRNA and protein expression. Exogenously expressed MCV large T antigen increased survivin protein expression in non-MCC primary cells. This required an intact retinoblastoma protein-targeting domain that activated survivin gene transcription as well as expression of other G(1)-S-phase proteins including E2F1 and cyclin E. Survivin expression is critical to the survival of MCV-positive MCC cells. A small-molecule survivin inhibitor, YM155, potently and selectively initiates irreversible, nonapoptotic, programmed MCV-positive MCC cell death. Of 1360 other chemotherapeutic and pharmacologically active compounds screened in vitro, only bortezomib (Velcade) was found to be similarly potent, but was not selective in killing MCV-positive MCC cells. YM155 halted the growth of MCV-positive MCC xenograft tumors and was nontoxic in mice, whereas bortezomib was not active in vivo and mice displayed serious morbidity. Xenograft tumors resumed growth once YM155 treatment was stopped, suggesting that YM155 may be cytostatic rather than cytotoxic in vivo. Identifying the cellular pathways, such as those involving survivin, that are targeted by tumor viruses can lead to rapid and rational identification of drug candidates for treating virus-induced cancers.
Conflict of interest statement
Figures


References
-
- Allen PJ, Bowne WB, Jaques DP, Brennan MF, Busam K, Coit DG. Merkel cell carcinoma: Prognosis and treatment of patients from a single institution. J Clin Oncol. 2005;23:2300–2309. - PubMed
-
- Veness M, Foote M, Gebski V, Poulsen M. The role of radiotherapy alone in patients with Merkel cell carcinoma: Reporting the Australian experience of 43 patients. Int J Radiat Oncol Biol Phys. 2010;78:703–709. - PubMed
-
- Eng TY, Boersma MG, Fuller CD, Goytia V, Jones WE, III, Joyner M, Nguyen DD. A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol. 2007;30:624–636. - PubMed
-
- Lemos B, Nghiem P. Merkel cell carcinoma: More deaths but still no pathway to blame. J Invest Dermatol. 2007;127:2100–2103. - PubMed
-
- Hodgson NC. Merkel cell carcinoma: Changing incidence trends. J Surg Oncol. 2005;89:1–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
