

Two protein kinase C isoforms, δ and ε, regulate energy homeostasis in mitochondria by transmitting opposing signals to the pyruvate dehydrogenase complex
- PMID: 22573912
- PMCID: PMC3405274
- DOI: 10.1096/fj.11-197376
Two protein kinase C isoforms, δ and ε, regulate energy homeostasis in mitochondria by transmitting opposing signals to the pyruvate dehydrogenase complex
Abstract
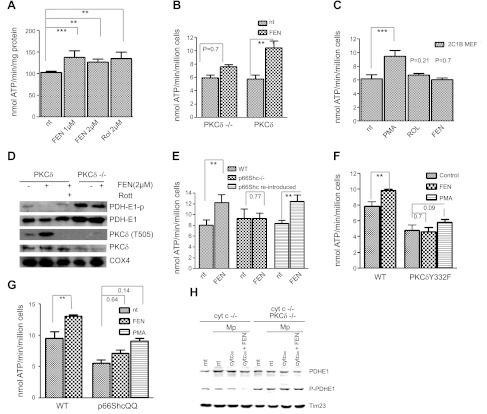
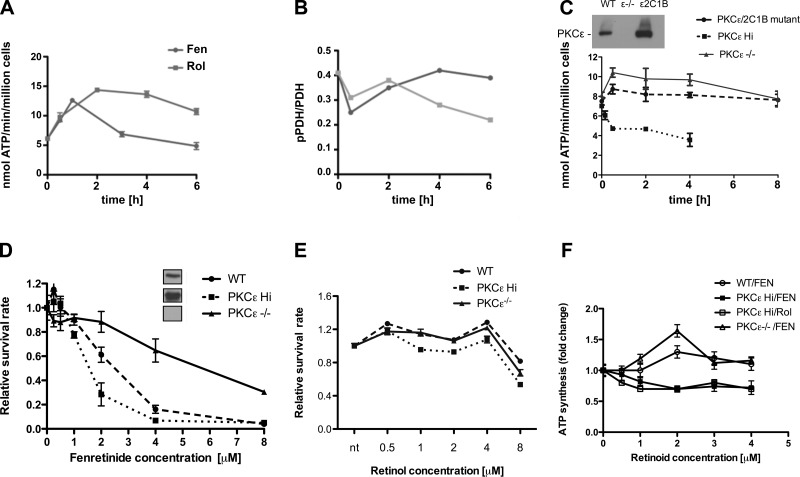
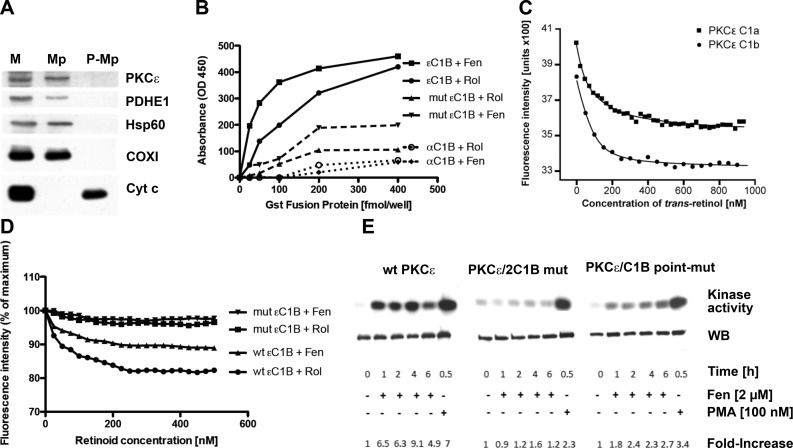
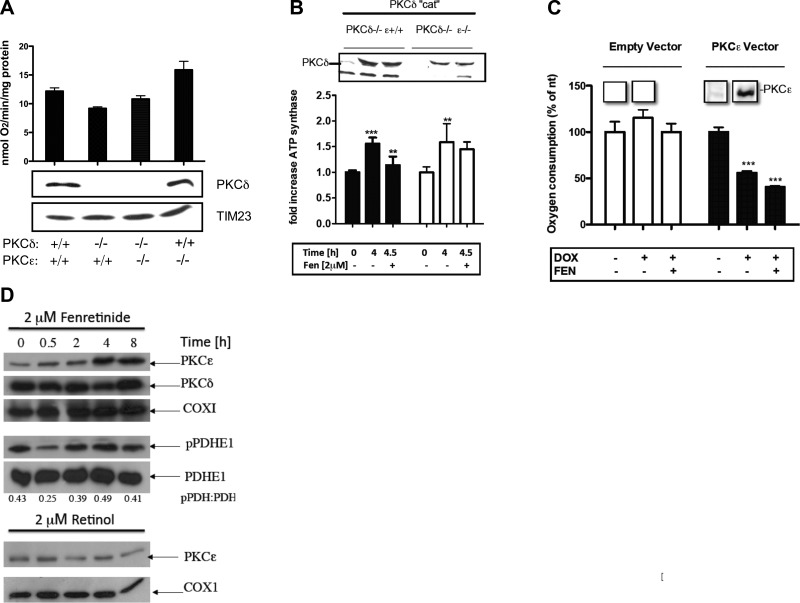
Energy production in mitochondria is a multistep process that requires coordination of several subsystems. While reversible phosphorylation is emerging as the principal tool, it is still unclear how this signal network senses the workloads of processes as different as fuel procurement, catabolism in the Krebs cycle, and stepwise oxidation of reducing equivalents in the electron transfer chain. We previously proposed that mitochondria use oxidized cytochrome c in concert with retinol to activate protein kinase Cδ, thereby linking a prominent kinase network to the redox balance of the ETC. Here, we show that activation of PKCε in mitochondria also requires retinol as a cofactor, implying a redox-mechanism. Whereas activated PKCδ transmits a stimulatory signal to the pyruvate dehdyrogenase complex (PDHC), PKCε opposes this signal and inhibits the PDHC. Our results suggest that the balance between PKCδ and ε is of paramount importance not only for flux of fuel entering the Krebs cycle but for overall energy homeostasis. We observed that the synthetic retinoid fenretinide substituted for the retinol cofactor function but, on chronic use, distorted this signal balance, leading to predominance of PKCε over PKCδ. The suppression of the PDHC might explain the proapoptotic effect of fenretinide on tumor cells, as well as the diminished adiposity observed in experimental animals and humans. Furthermore, a disturbed balance between PKCδ and PKCε might underlie the injury inflicted on the ischemic myocardium during reperfusion. dehydrogenase complex.
Figures
References
-
- Holness M. J., Sugden M. C. (2003) Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 31, 1143–1151 - PubMed
-
- Huang B., Gudi R., Wu P., Harris R. A., Hamilton J., Popov K. M. (1998) Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J. Biol. Chem. 273, 17680–17688 - PubMed
-
- D'Aurelio M., Pallotti F., Barrientos A., Gajewski C. D., Kwong J. Q., Bruno C., Beal M. F., Manfredi G. (2001) In vivo regulation of oxidative phosphorylation in cells harboring a stop-codon mutation in mitochondrial DNA-encoded cytochrome c oxidase subunit I. J. Biol. Chem. 276, 46925–46932 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
