

Improving ancient DNA read mapping against modern reference genomes
- PMID: 22574660
- PMCID: PMC3468387
- DOI: 10.1186/1471-2164-13-178
Improving ancient DNA read mapping against modern reference genomes
Abstract
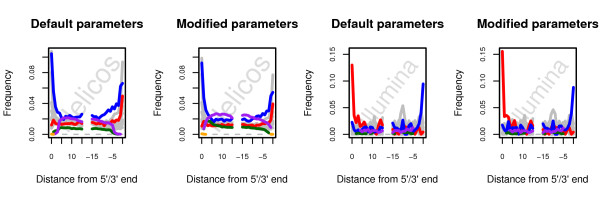
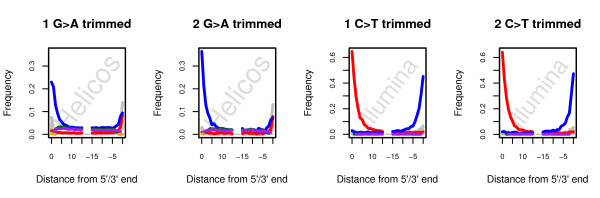
Background: Next-Generation Sequencing has revolutionized our approach to ancient DNA (aDNA) research, by providing complete genomic sequences of ancient individuals and extinct species. However, the recovery of genetic material from long-dead organisms is still complicated by a number of issues, including post-mortem DNA damage and high levels of environmental contamination. Together with error profiles specific to the type of sequencing platforms used, these specificities could limit our ability to map sequencing reads against modern reference genomes and therefore limit our ability to identify endogenous ancient reads, reducing the efficiency of shotgun sequencing aDNA.
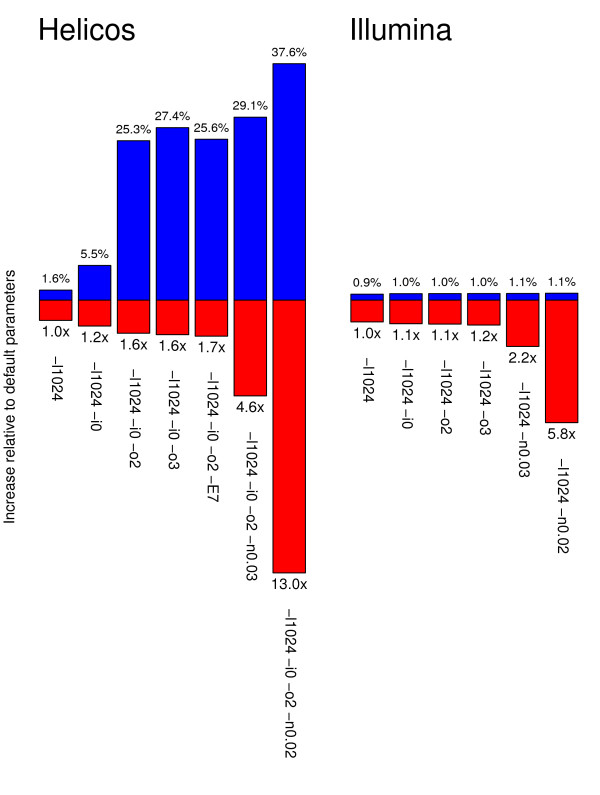
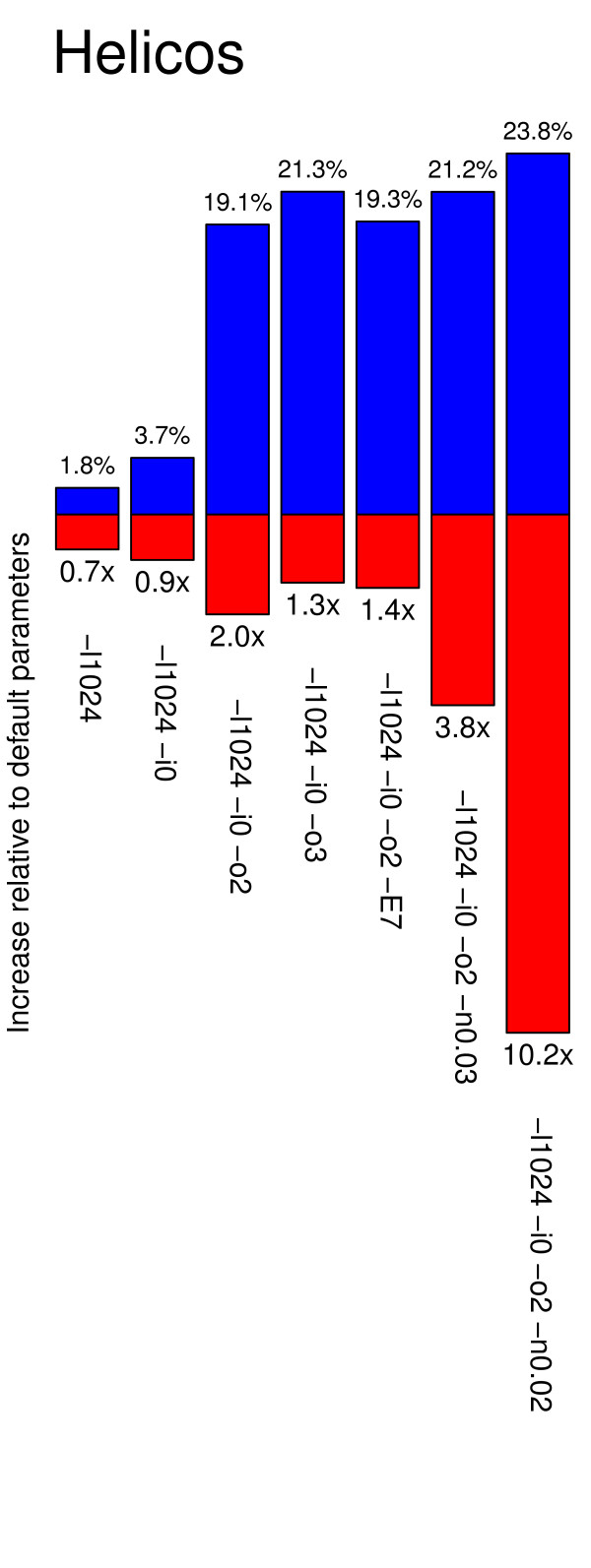
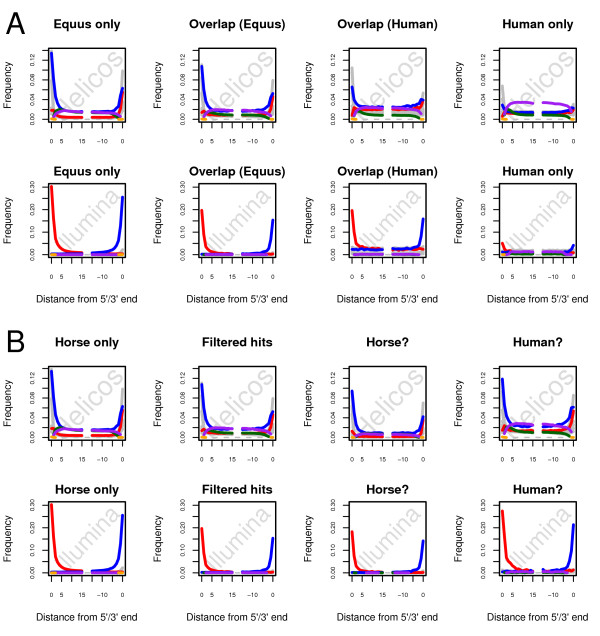
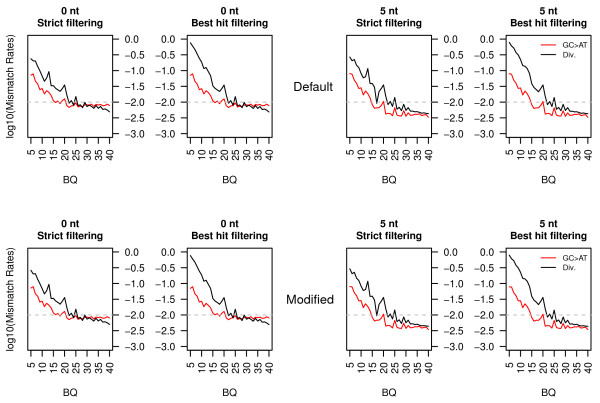
Results: In this study, we compare different computational methods for improving the accuracy and sensitivity of aDNA sequence identification, based on shotgun sequencing reads recovered from Pleistocene horse extracts using Illumina GAIIx and Helicos Heliscope platforms. We show that the performance of the Burrows Wheeler Aligner (BWA), that has been developed for mapping of undamaged sequencing reads using platforms with low rates of indel-types of sequencing errors, can be employed at acceptable run-times by modifying default parameters in a platform-specific manner. We also examine if trimming likely damaged positions at read ends can increase the recovery of genuine aDNA fragments and if accurate identification of human contamination can be achieved using a strategy previously suggested based on best hit filtering. We show that combining our different mapping and filtering approaches can increase the number of high-quality endogenous hits recovered by up to 33%.
Conclusions: We have shown that Illumina and Helicos sequences recovered from aDNA extracts could not be aligned to modern reference genomes with the same efficiency unless mapping parameters are optimized for the specific types of errors generated by these platforms and by post-mortem DNA damage. Our findings have important implications for future aDNA research, as we define mapping guidelines that improve our ability to identify genuine aDNA sequences, which in turn could improve the genotyping accuracy of ancient specimens. Our framework provides a significant improvement to the standard procedures used for characterizing ancient genomes, which is challenged by contamination and often low amounts of DNA material.
Figures

References
-
- Willerslev E, Cappellini E, Boomsma W, Nielsen R, Hebsgaard MB, Brand TB, Hofreiter M, Bunce M, Poinar HN, Dahl-Jensen D, Johnsen S, Steffensen JP, Bennike O, Schwenninger J-L, Nathan R, Armitage S, de Hoog C-J, Alfimov V, Christl M, Beer J, Muscheler R, Barker J, Sharp M, Penkman KEH, Haile J, Taberlet P, Gilbert MTP, Casoli A, Campani E, Collins MJ. Ancient biomolecules from deep ice cores reveal a forested southern Greenland. Science. 2007;317:111–114. doi: 10.1126/science.1141758. - DOI - PMC - PubMed
-
- Stiller M, Baryshnikov G, Bocherens H, AG d’Anglade null, Hilpert B, Münzel SC, Pinhasi R, Rabeder G, Rosendahl W, Trinkaus E, Hofreiter M, Knapp M. Withering away--25,000 years of genetic decline preceded cave bear extinction. Mol Biol Evol. 2010;27:975–978. doi: 10.1093/molbev/msq083. - DOI - PubMed
-
- Lorenzen ED, Nogués-Bravo D, Orlando L, Weinstock J, Binladen J, Marske KA, Ugan A, Borregaard MK, Gilbert MTP, Nielsen R, Ho SYW, Goebel T, Graf KE, Byers D, Stenderup JT, Rasmussen M, Campos PF, Leonard JA, Koepfli K-P, Froese D, Zazula G, Stafford TW, Aaris-Sørensen K, Batra P, Haywood AM, Singarayer JS, Valdes PJ, Boeskorov G, Burns JA, Davydov SP, Haile J, Jenkins DL, Kosintsev P, Kuznetsova T, Lai X, Martin LD, McDonald HG, Mol D, Meldgaard M, Munch K, Stephan E, Sablin M, Sommer RS, Sipko T, Scott E, Suchard MA, Tikhonov A, Willerslev R, Wayne RK, Cooper A, Hofreiter M, Sher A, Shapiro B, Rahbek C, Willerslev E. Species-specific responses of Late Quaternary megafauna to climate and humans. Nature. 2011;479:359–364. doi: 10.1038/nature10574. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
