

Disease severity in a mouse model of ataxia telangiectasia is modulated by the DNA damage checkpoint gene Hus1
- PMID: 22575700
- PMCID: PMC3392115
- DOI: 10.1093/hmg/dds173
Disease severity in a mouse model of ataxia telangiectasia is modulated by the DNA damage checkpoint gene Hus1
Abstract
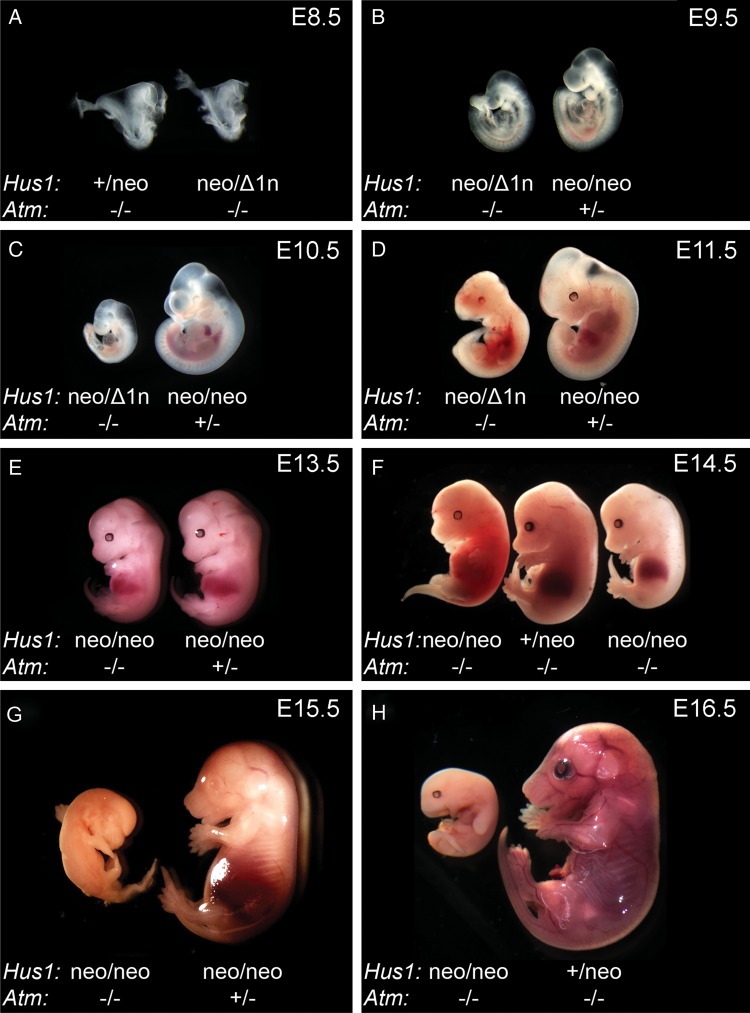
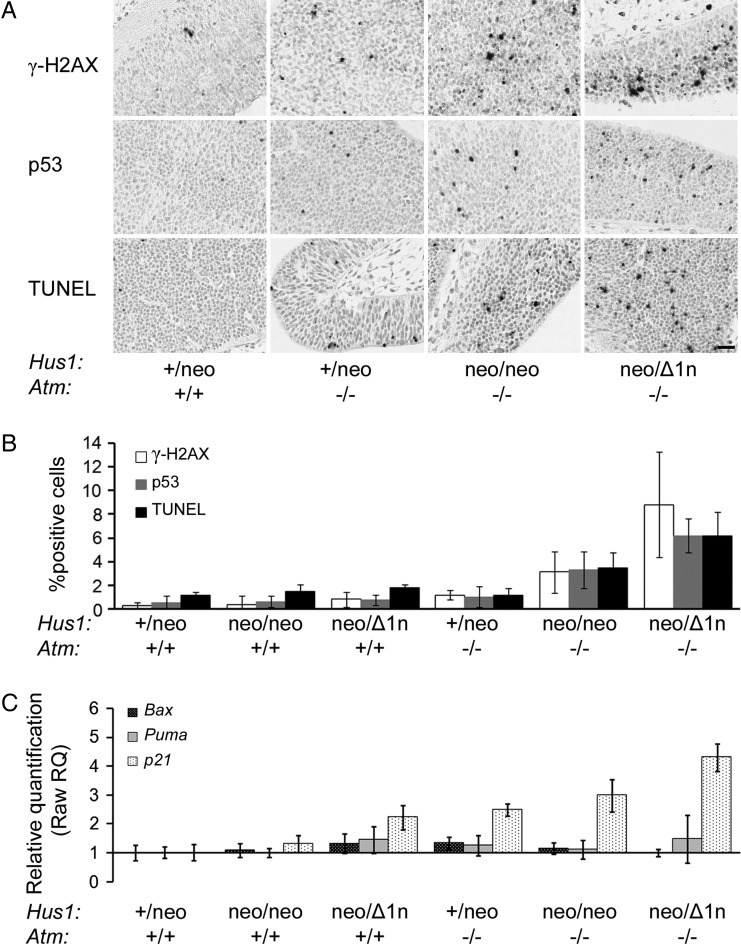
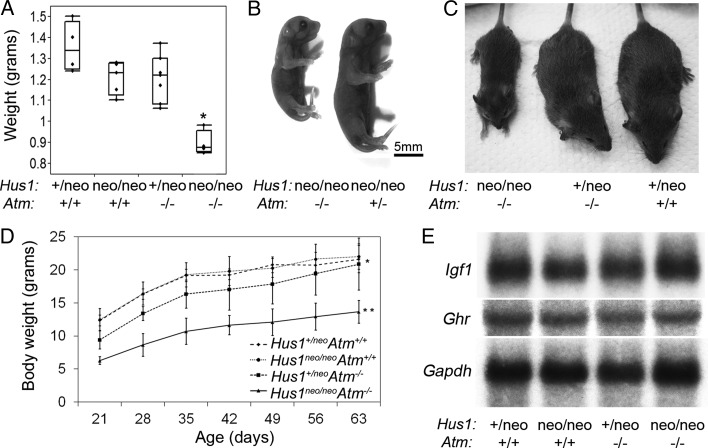
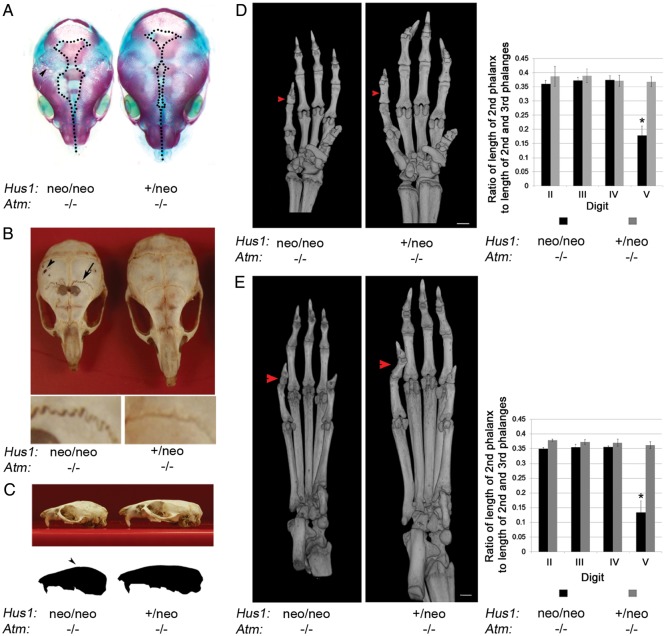
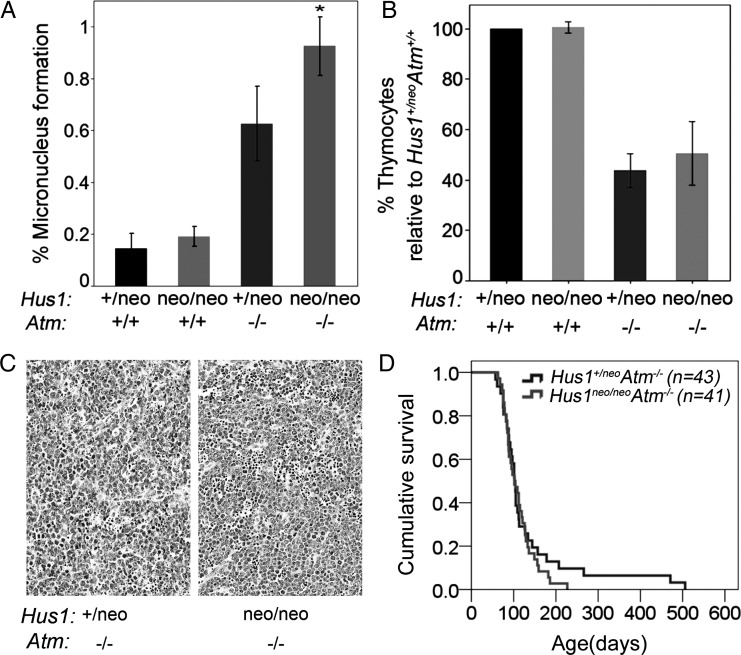
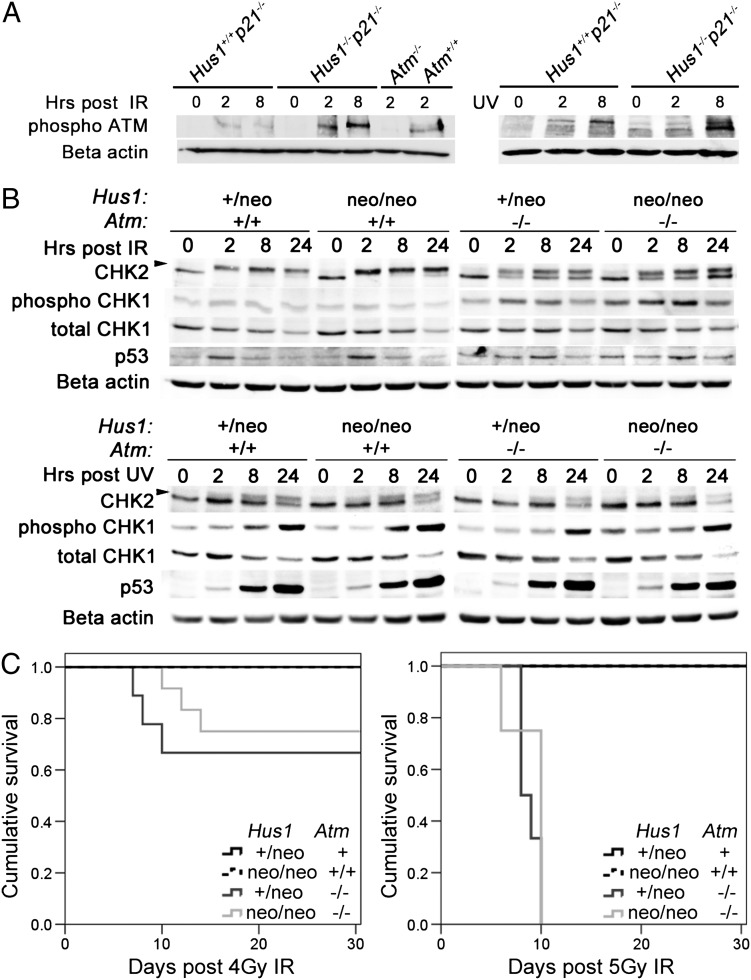
The human genomic instability syndrome ataxia telangiectasia (A-T), caused by mutations in the gene encoding the DNA damage checkpoint kinase ATM, is characterized by multisystem defects including neurodegeneration, immunodeficiency and increased cancer predisposition. ATM is central to a pathway that responds to double-strand DNA breaks, whereas the related kinase ATR leads a parallel signaling cascade that is activated by replication stress. To dissect the physiological relationship between the ATM and ATR pathways, we generated mice defective for both. Because complete ATR pathway inactivation causes embryonic lethality, we weakened the ATR mechanism to different degrees by impairing HUS1, a member of the 911 complex that is required for efficient ATR signaling. Notably, simultaneous ATM and HUS1 defects caused synthetic lethality. Atm/Hus1 double-mutant embryos showed widespread apoptosis and died mid-gestationally. Despite the underlying DNA damage checkpoint defects, increased DNA damage signaling was observed, as evidenced by H2AX phosphorylation and p53 accumulation. A less severe Hus1 defect together with Atm loss resulted in partial embryonic lethality, with the surviving double-mutant mice showing synergistic increases in genomic instability and specific developmental defects, including dwarfism, craniofacial abnormalities and brachymesophalangy, phenotypes that are observed in several human genomic instability disorders. In addition to identifying tissue-specific consequences of checkpoint dysfunction, these data highlight a robust, cooperative configuration for the mammalian DNA damage response network and further suggest HUS1 and related genes in the ATR pathway as candidate modifiers of disease severity in A-T patients.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
