

Structural and energetic basis of allostery
- PMID: 22577828
- PMCID: PMC3935618
- DOI: 10.1146/annurev-biophys-050511-102319
Structural and energetic basis of allostery
Abstract
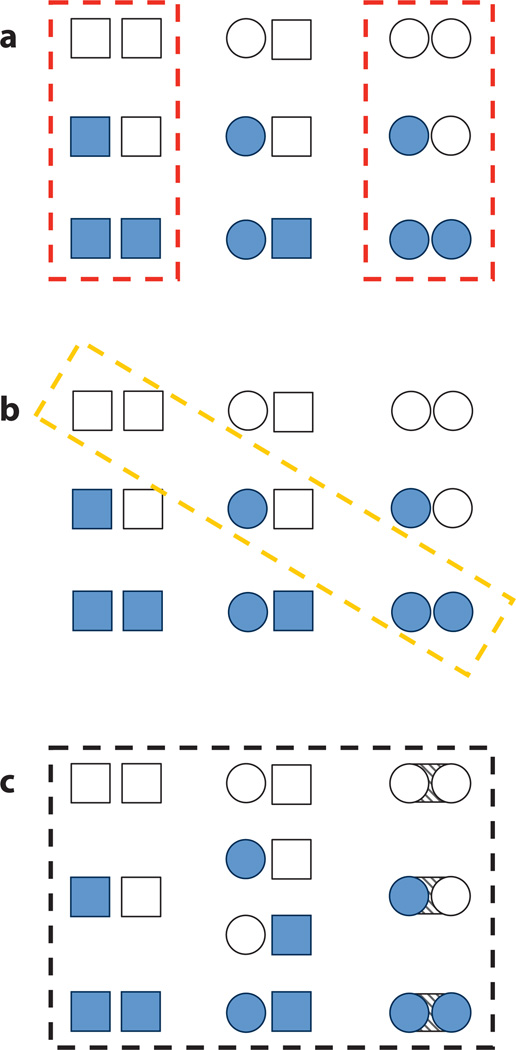
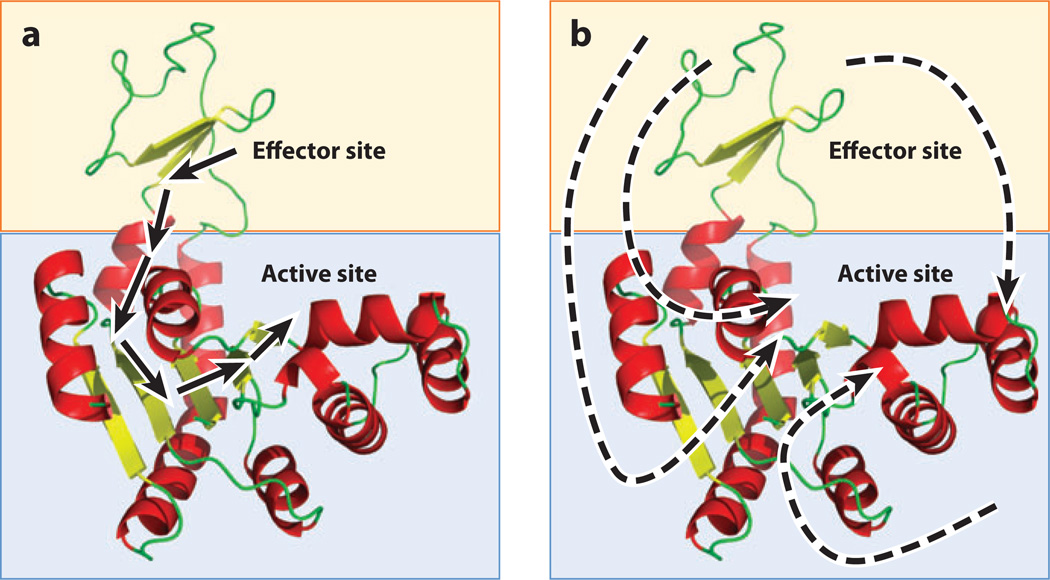
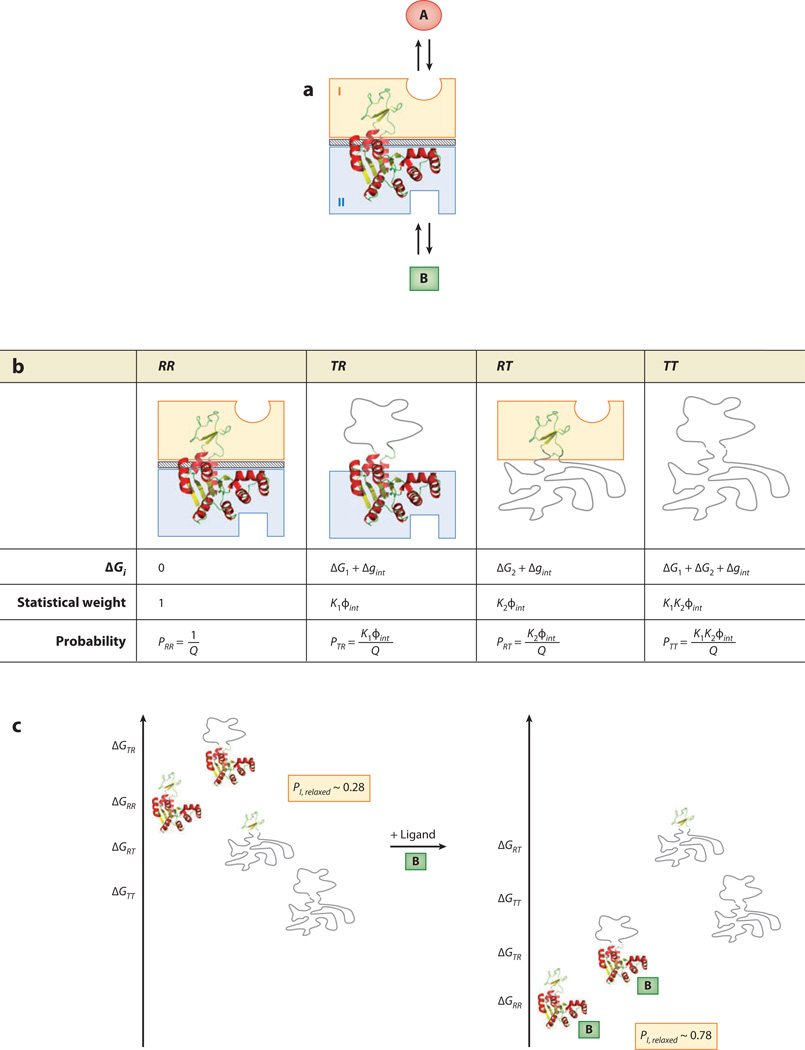
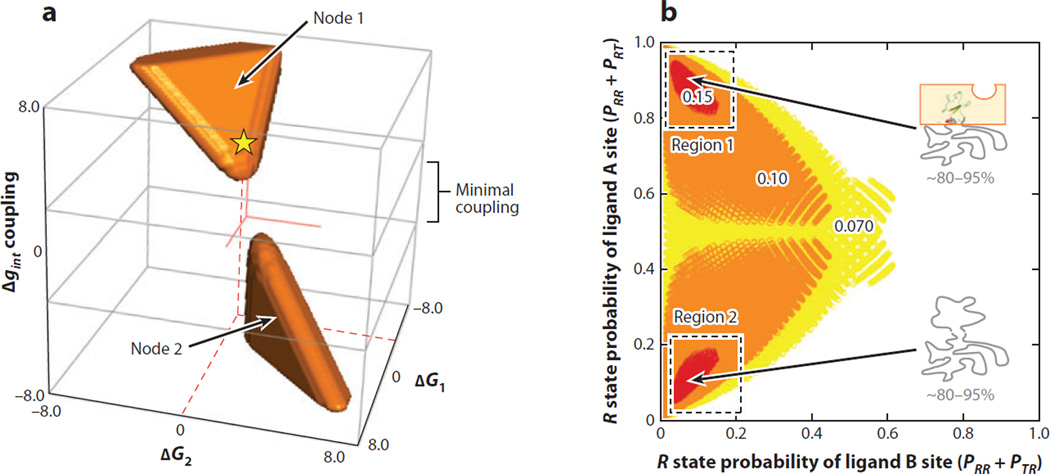
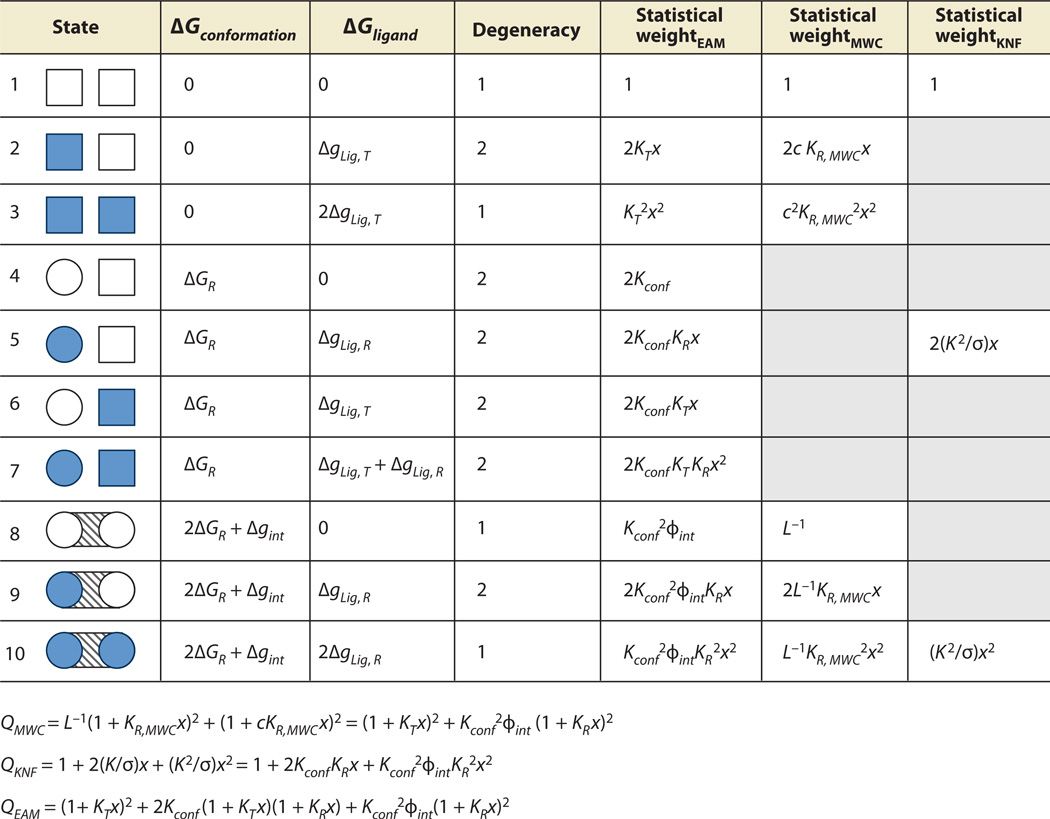
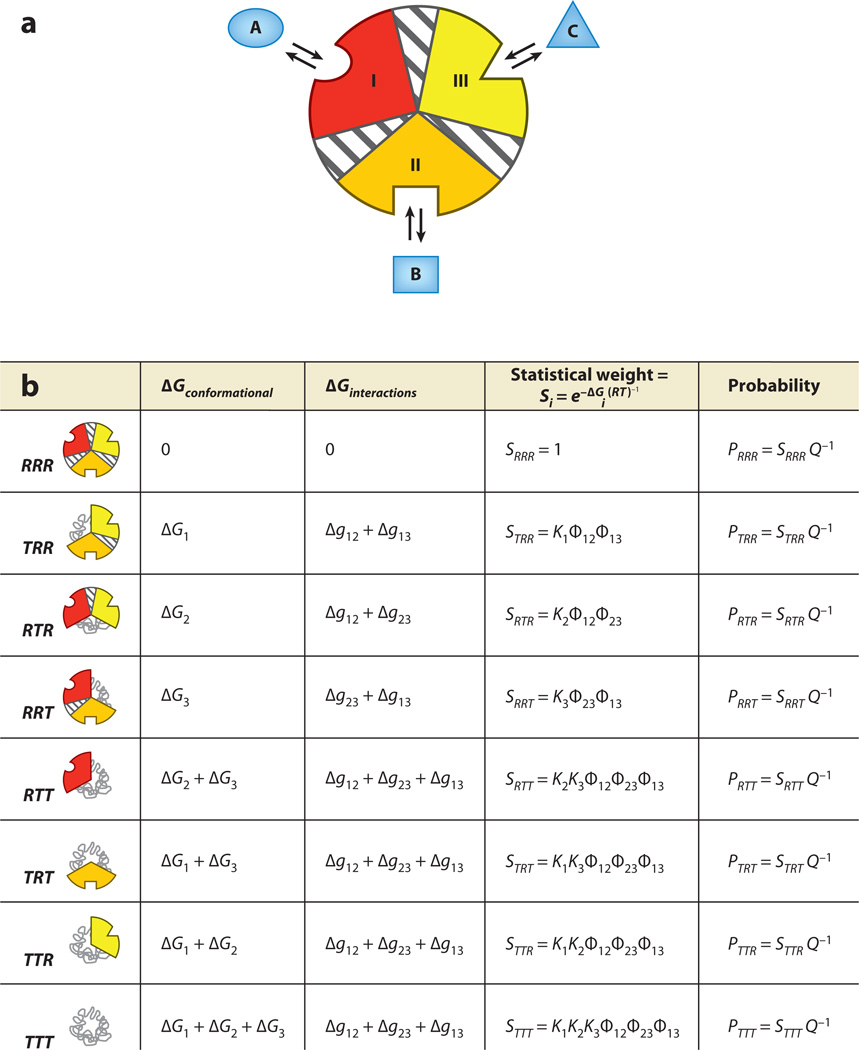
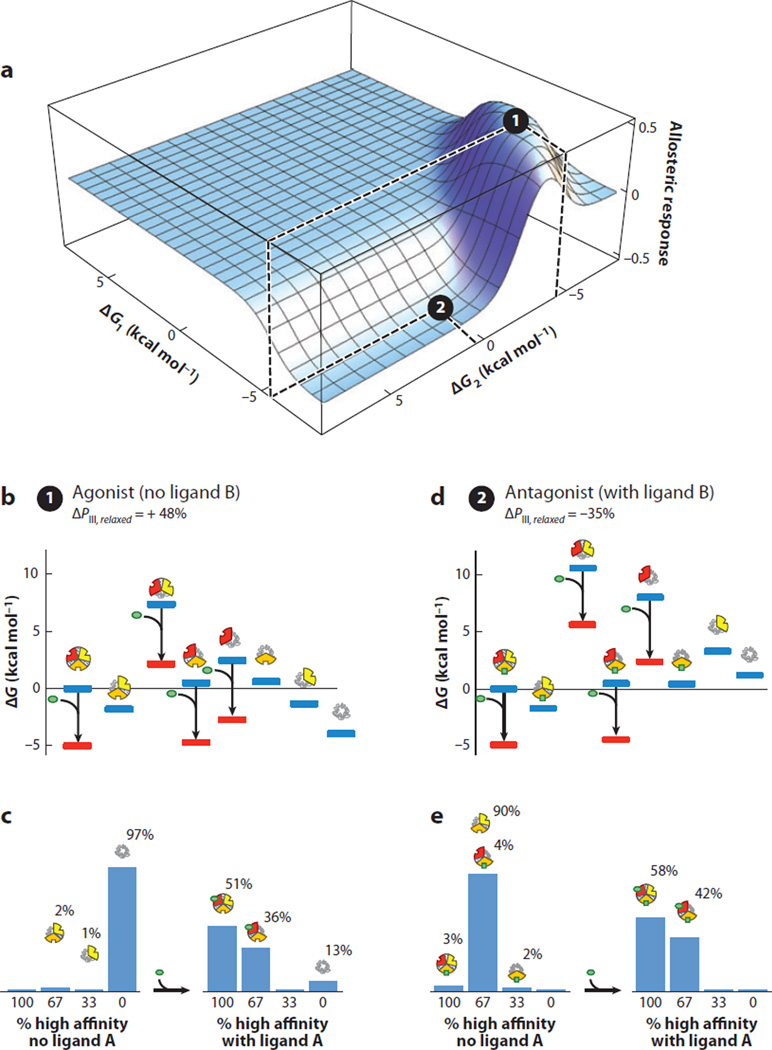
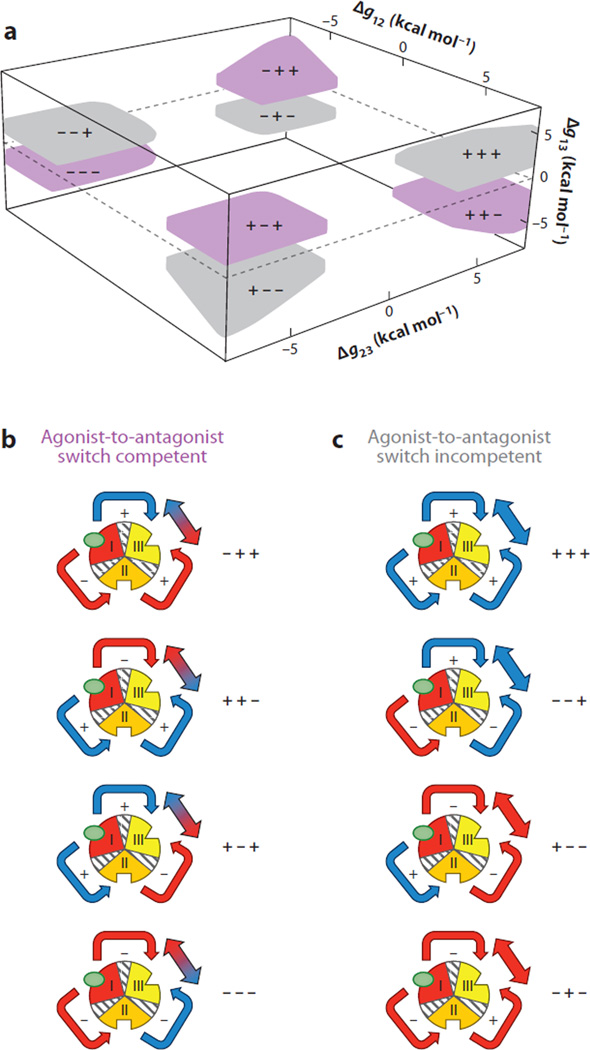
Allostery is a biological phenomenon of fundamental importance in regulation and signaling, and efforts to understand this process have led to the development of numerous models. In spite of individual successes in understanding the structural determinants of allostery in well-documented systems, much less success has been achieved in identifying a set of quantitative and transferable ground rules that provide an understanding of how allostery works. Are there organizing principles that allow us to relate structurally different proteins, or are the determinants of allostery unique to each system? Using an ensemble-based model, we show that allosteric phenomena can be formulated in terms of conformational free energies of the cooperative elements in a protein and the coupling interactions between them. Interestingly, the resulting allosteric ground rules provide a framework to reconcile observations that challenge purely structural models of site-to-site coupling, including (a) allostery in the absence of pathways of structural distortions, (b) allostery in the absence of any structural change, and (c) the ability of allosteric ligands to act as agonists under some circumstances and antagonists under others. The ensemble view of allostery that emerges provides insights into the energetic prerequisites of site-to-site coupling and thus into how allostery works.
Figures
References
-
- Branden C, Tooze J. Introduction to Protein Structure. New York: Garland Science; 1999.
-
- Cesareni G, Gimona M, Sudol M, Yaffe M, editors. Modular Protein Domains. Weinheim FRG: Wiley-VCH; 2005.
-
- Changeux JP. The feedback control mechanisms of biosynthetic L-threonine deaminase by L-isoleucine. Cold Spring Harbor Symp. Quant. Biol. 1961;26:313–318. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
