

GPSM2 mutations cause the brain malformations and hearing loss in Chudley-McCullough syndrome
- PMID: 22578326
- PMCID: PMC3370271
- DOI: 10.1016/j.ajhg.2012.04.008
GPSM2 mutations cause the brain malformations and hearing loss in Chudley-McCullough syndrome
Erratum in
- Am J Hum Genet. 2012 Jul 13;91(1):209
Abstract
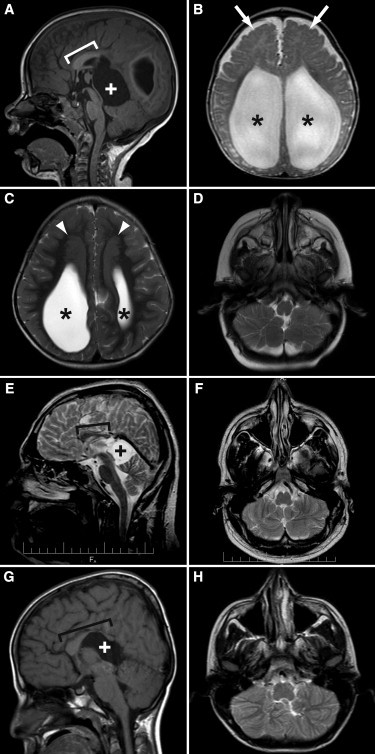
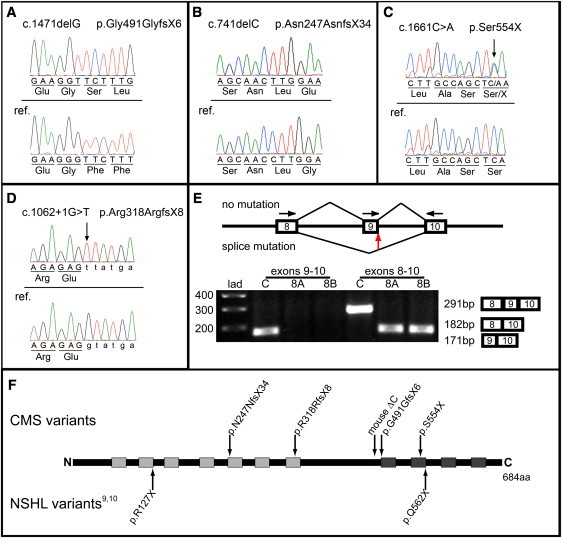
Autosomal-recessive inheritance, severe to profound sensorineural hearing loss, and partial agenesis of the corpus callosum are hallmarks of the clinically well-established Chudley-McCullough syndrome (CMS). Although not always reported in the literature, frontal polymicrogyria and gray matter heterotopia are uniformly present, whereas cerebellar dysplasia, ventriculomegaly, and arachnoid cysts are nearly invariant. Despite these striking brain malformations, individuals with CMS generally do not present with significant neurodevelopmental abnormalities, except for hearing loss. Homozygosity mapping and whole-exome sequencing of DNA from affected individuals in eight families (including the family in the first report of CMS) revealed four molecular variations (two single-base deletions, a nonsense mutation, and a canonical splice-site mutation) in the G protein-signaling modulator 2 gene, GPSM2, that underlie CMS. Mutations in GPSM2 have been previously identified in people with profound congenital nonsyndromic hearing loss (NSHL). Subsequent brain imaging of these individuals revealed frontal polymicrogyria, abnormal corpus callosum, and gray matter heterotopia, consistent with a CMS diagnosis, but no ventriculomegaly. The gene product, GPSM2, is required for orienting the mitotic spindle during cell division in multiple tissues, suggesting that the sensorineural hearing loss and characteristic brain malformations of CMS are due to defects in asymmetric cell divisions during development.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Chudley A.E., McCullough C., McCullough D.W. Bilateral sensorineural deafness and hydrocephalus due to foramen of Monro obstruction in sibs: a newly described autosomal recessive disorder. Am. J. Med. Genet. 1997;68:350–356. - PubMed
-
- Hendriks Y.M.C., Laan L.A.E.M., Vielvoye G.J., van Haeringen A. Bilateral sensorineural deafness, partial agenesis of the corpus callosum, and arachnoid cysts in two sisters. Am. J. Med. Genet. 1999;86:183–186. - PubMed
-
- Lemire E.G., Stoeber G.P. Chudley-McCullough syndrome: bilateral sensorineural deafness, hydrocephalus, and other structural brain abnormalities. Am. J. Med. Genet. 2000;90:127–130. - PubMed
-
- Welch K.O., Tekin M., Nance W.E., Blanton S.H., Arnos K.S., Pandya A. Chudley-McCullough syndrome: expanded phenotype and review of the literature. Am. J. Med. Genet. A. 2003;119A:71–76. - PubMed
-
- Østergaard E., Pedersen V.F., Skriver E.B., Brøndum-Nielsen K. Brothers with Chudley-McCullough syndrome: sensorineural deafness, agenesis of the corpus callosum, and other structural brain abnormalities. Am. J. Med. Genet. A. 2004;124A:74–78. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
