

Three-dimensional structures of membrane proteins from genomic sequencing
- PMID: 22579045
- PMCID: PMC3641781
- DOI: 10.1016/j.cell.2012.04.012
Three-dimensional structures of membrane proteins from genomic sequencing
Abstract
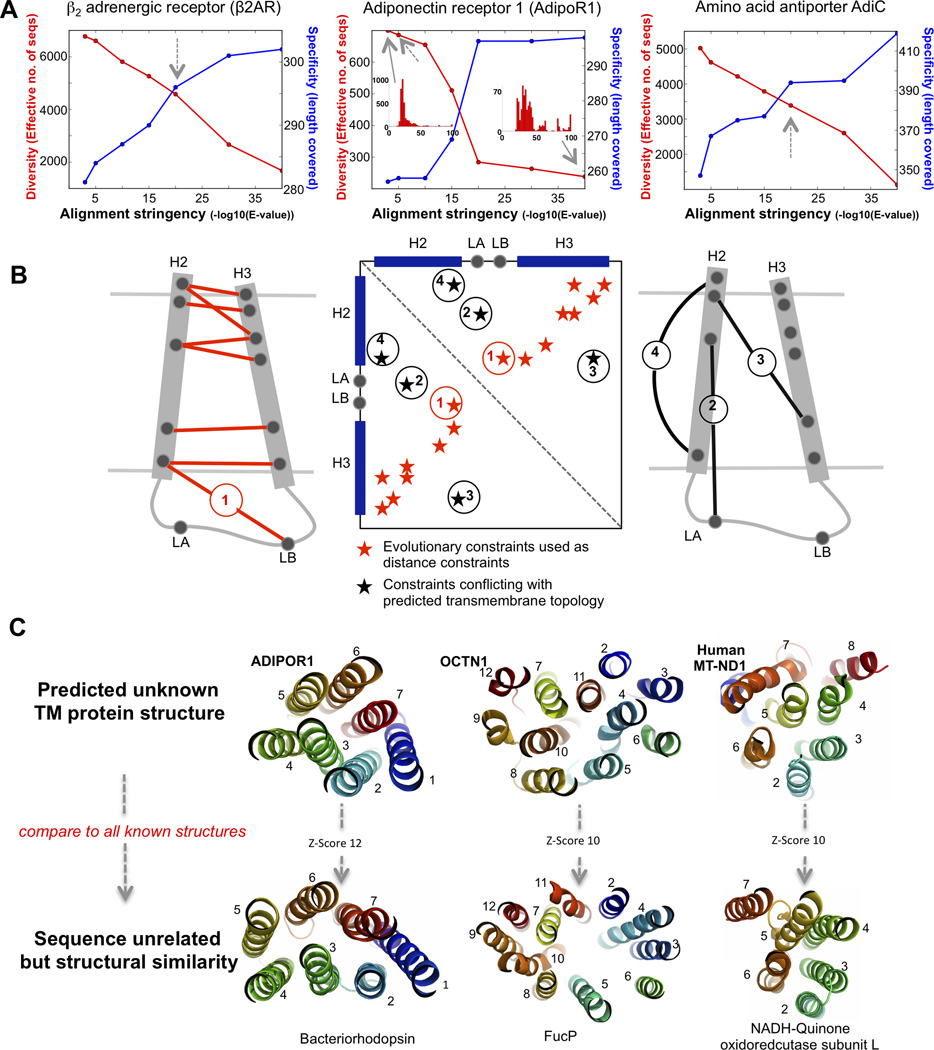
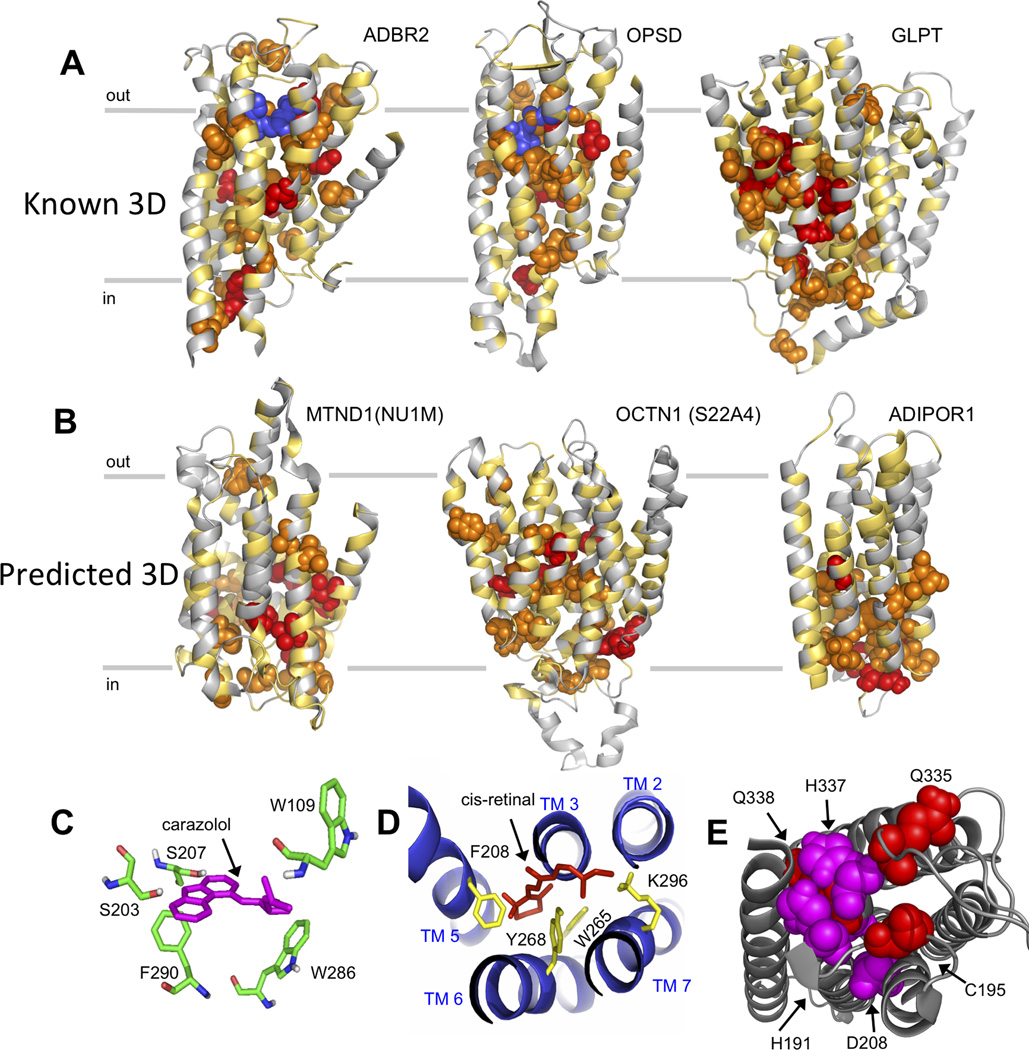
We show that amino acid covariation in proteins, extracted from the evolutionary sequence record, can be used to fold transmembrane proteins. We use this technique to predict previously unknown 3D structures for 11 transmembrane proteins (with up to 14 helices) from their sequences alone. The prediction method (EVfold_membrane) applies a maximum entropy approach to infer evolutionary covariation in pairs of sequence positions within a protein family and then generates all-atom models with the derived pairwise distance constraints. We benchmark the approach with blinded de novo computation of known transmembrane protein structures from 23 families, demonstrating unprecedented accuracy of the method for large transmembrane proteins. We show how the method can predict oligomerization, functional sites, and conformational changes in transmembrane proteins. With the rapid rise in large-scale sequencing, more accurate and more comprehensive information on evolutionary constraints can be decoded from genetic variation, greatly expanding the repertoire of transmembrane proteins amenable to modeling by this method.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures




Comment in
-
Membrane protein structure predictions for exploration.Cell. 2012 Jun 22;149(7):1424-5. doi: 10.1016/j.cell.2012.06.004. Cell. 2012. PMID: 22726429 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
