

Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion
- PMID: 22580601
- PMCID: PMC3592945
- DOI: 10.1038/onc.2012.148
Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion
Abstract
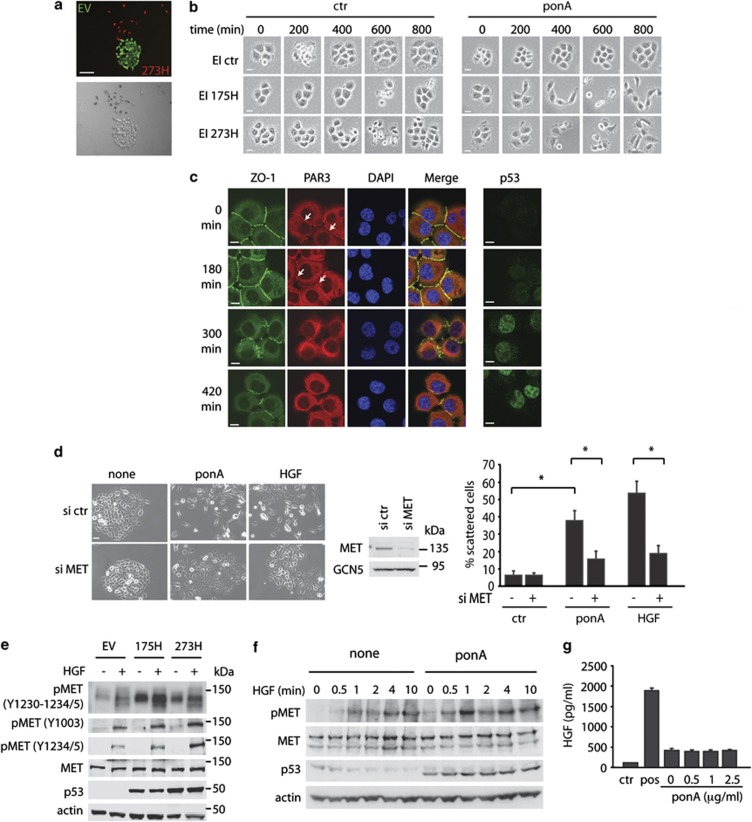
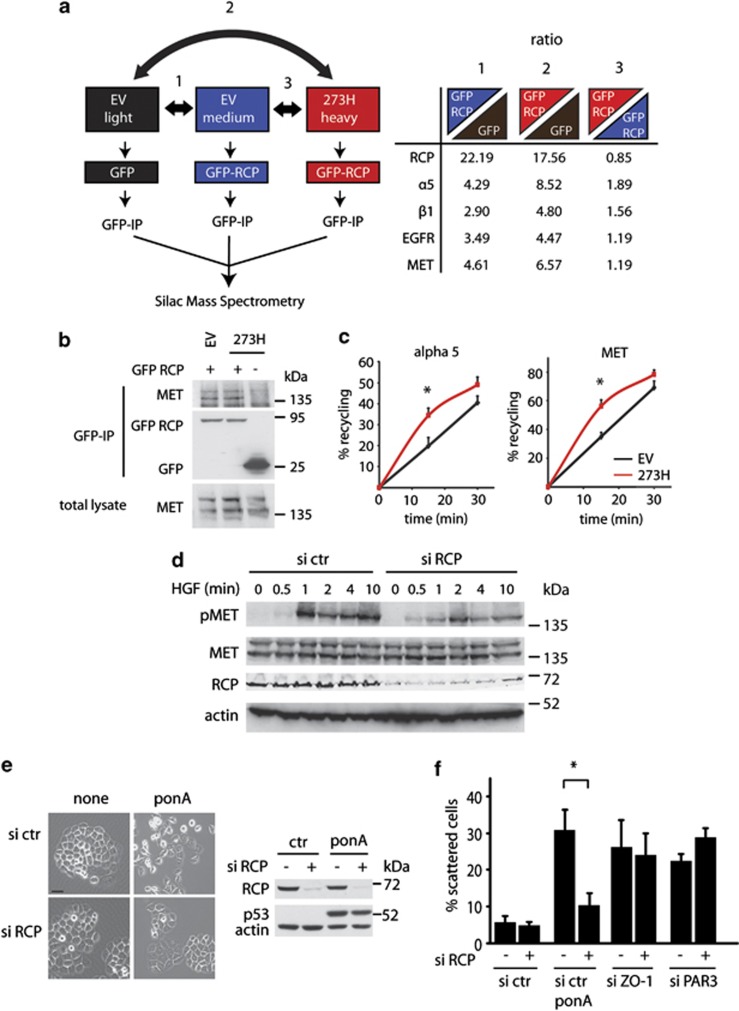
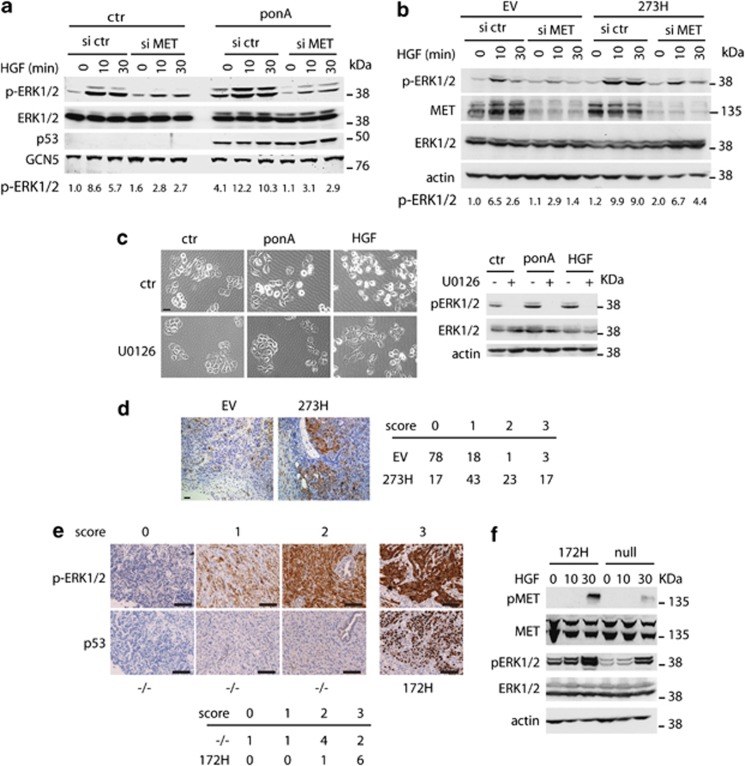
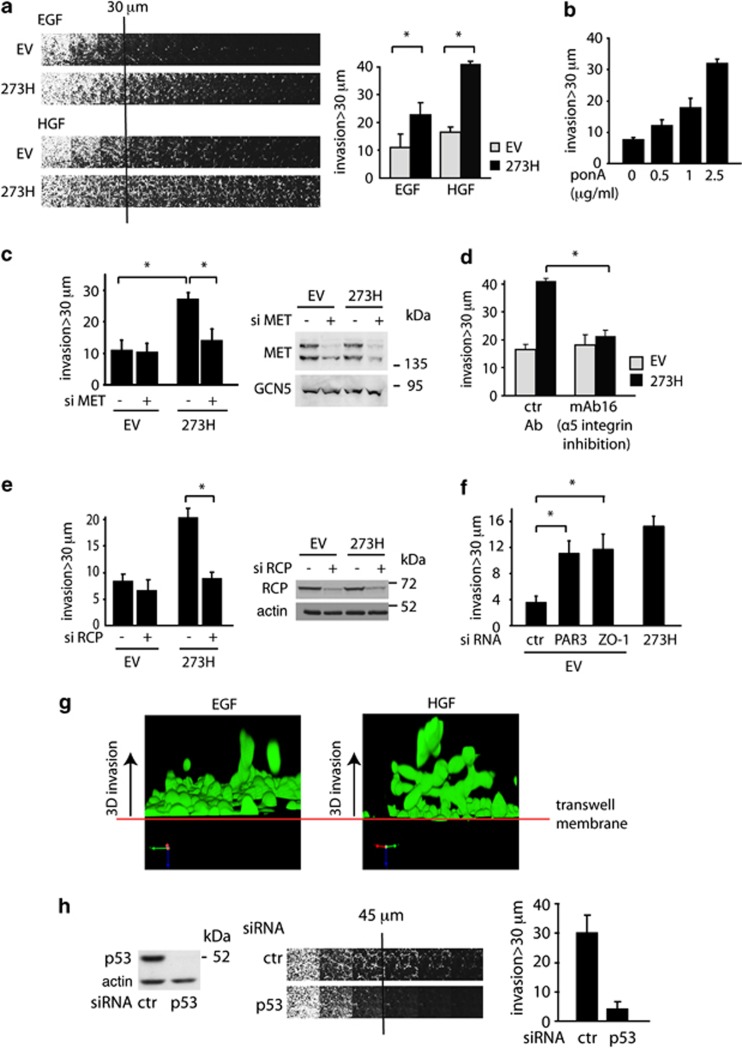
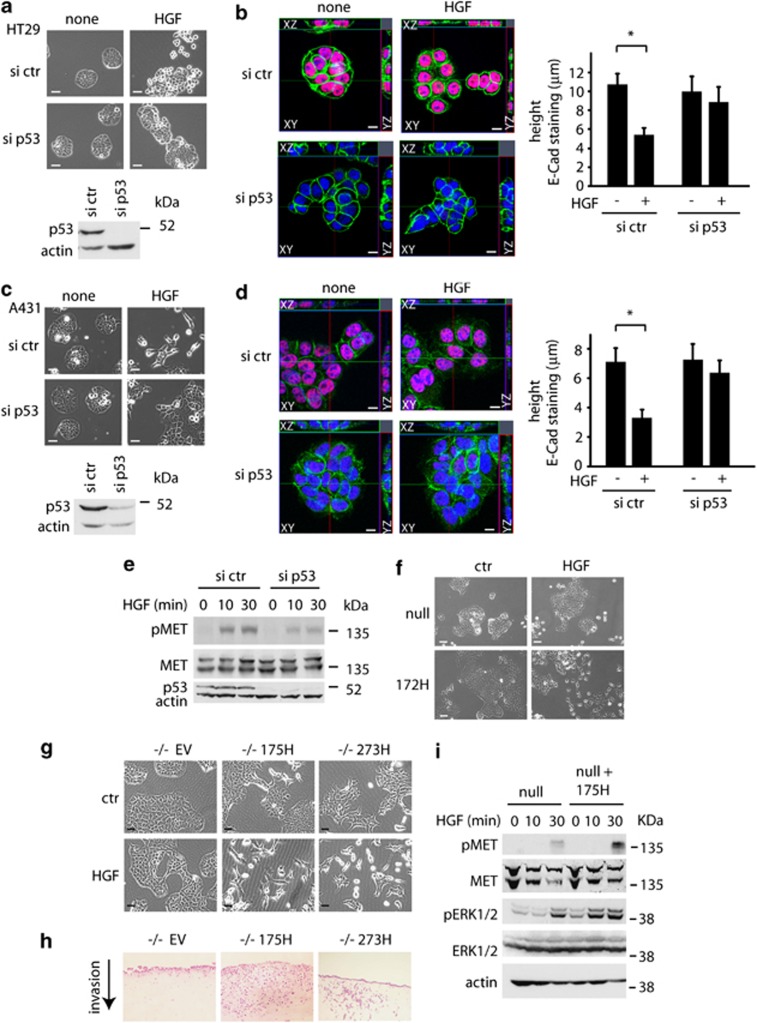
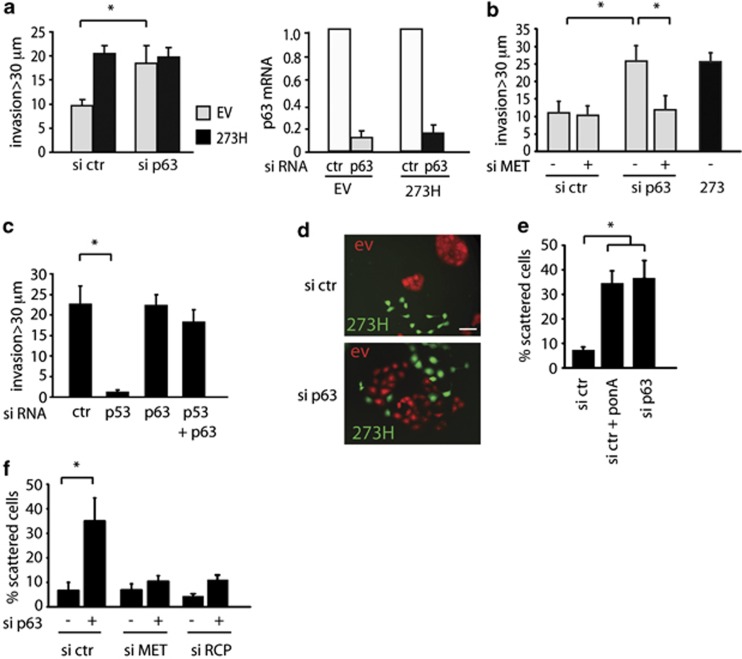
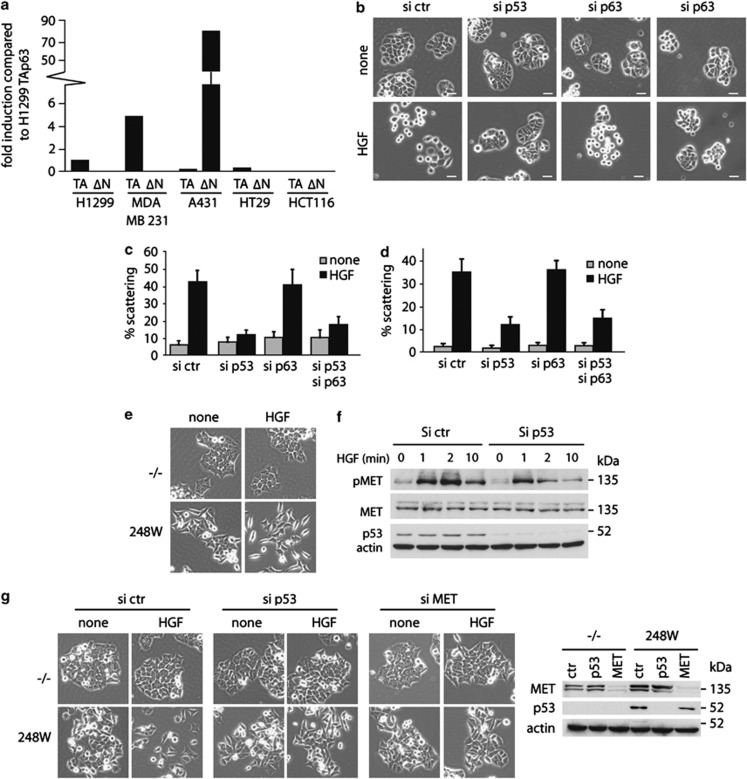
Tumour-derived mutant p53 proteins promote invasion, in part, by enhancing Rab coupling protein (RCP)-dependent receptor recycling. Here we identified MET as an RCP-binding protein and showed that mutant p53 promoted MET recycling. Mutant p53-expressing cells were more sensitive to hepatocyte growth factor, the ligand for MET, leading to enhanced MET signalling, invasion and cell scattering that was dependent on both MET and RCP. In cells expressing the p53 family member TAp63, inhibition of TAp63 also lead to cell scattering and MET-dependent invasion. However, in cells that express very low levels of TAp63, the ability of mutant p53 to promote MET-dependent cell scattering was independent of TAp63. Taken together, our data show that mutant p53 can enhance MET signalling to promote cell scattering and invasion through both TAp63-dependent and -independent mechanisms. MET has a predominant role in metastatic progression and the identification of mechanisms through which mutations in p53 can drive MET signalling may help to identify and direct therapy.
Figures
References
-
- Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 2000;60:6788–6793. - PubMed
-
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. - PubMed
-
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. - PubMed
-
- O'Farrell TJ, Ghosh P, Dobashi N, Sasaki CY, Longo DL. Comparison of the effect of mutant and wild-type p53 on global gene expression. Cancer Res. 2004;64:8199–8207. - PubMed
-
- Tepper CG, Gregg JP, Shi XB, Vinall RL, Baron CA, Ryan PE, et al. Profiling of gene expression changes caused by p53 gain-of-function mutant alleles in prostate cancer cells. Prostate. 2005;65:375–389. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
