

Downregulation of the tumor-suppressor miR-16 via progestin-mediated oncogenic signaling contributes to breast cancer development
- PMID: 22583478
- PMCID: PMC3446340
- DOI: 10.1186/bcr3187
Downregulation of the tumor-suppressor miR-16 via progestin-mediated oncogenic signaling contributes to breast cancer development
Abstract
Introduction: Experimental and clinical evidence points to a critical role of progesterone and the nuclear progesterone receptor (PR) in controlling mammary gland tumorigenesis. However, the molecular mechanisms of progesterone action in breast cancer still remain elusive. On the other hand, micro RNAs (miRNAs) are short ribonucleic acids which have also been found to play a pivotal role in cancer pathogenesis. The role of miRNA in progestin-induced breast cancer is poorly explored. In this study we explored progestin modulation of miRNA expression in mammary tumorigenesis.
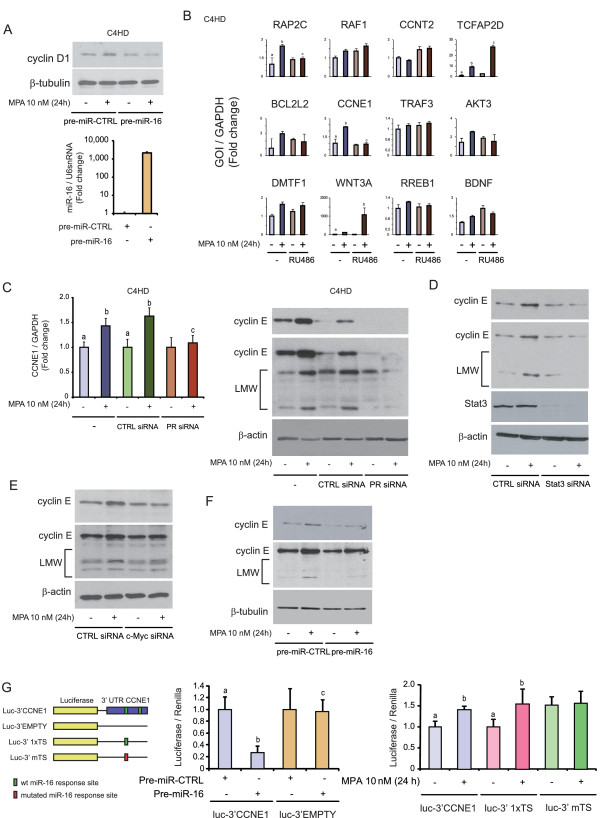
Methods: We performed a genome-wide study to explore progestin-mediated regulation of miRNA expression in breast cancer. miR-16 expression was studied by RT-qPCR in cancer cell lines with silenced PR, signal transducer and activator of transcription 3 (Stat3) or c-Myc, treated or not with progestins. Breast cancer cells were transfected with the precursor of miR-16 and proliferation assays, Western blots or in vivo experiments were performed. Target genes of miR-16 were searched through a bioinformatical approach, and the study was focused on cyclin E. Reporter gene assays were performed to confirm that cyclin E 3'UTR is a direct target of miR-16.
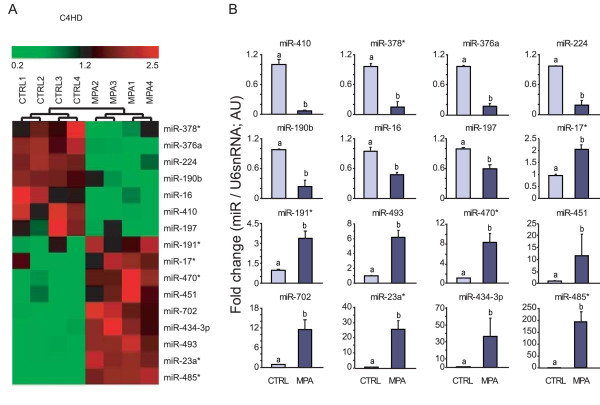
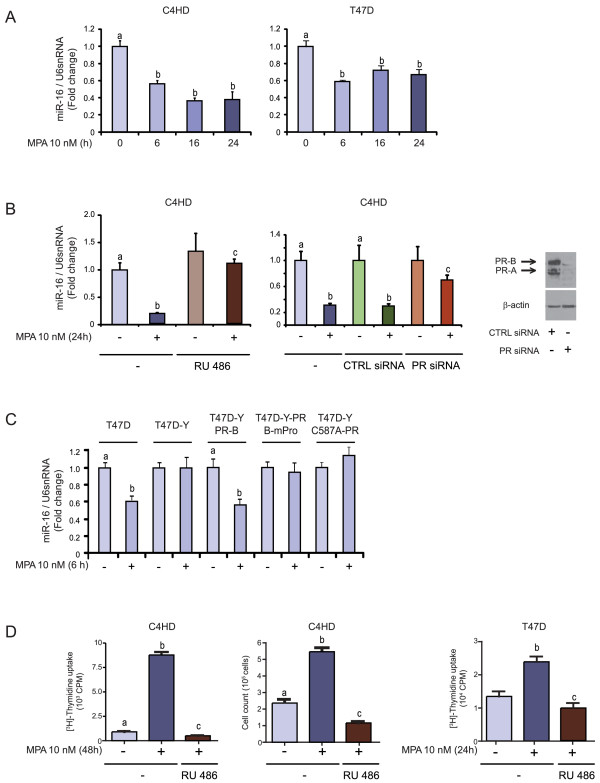
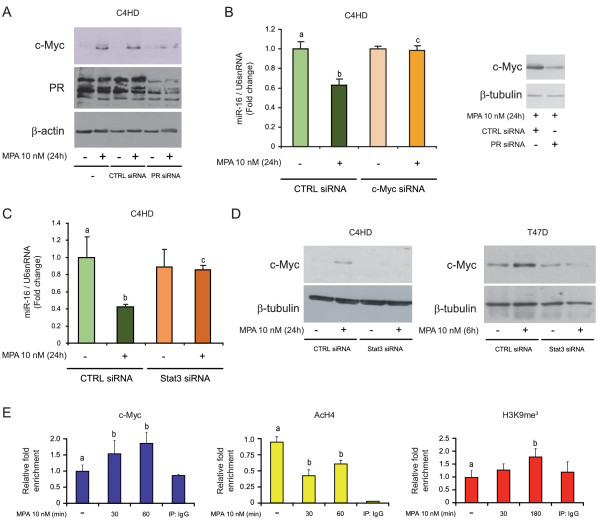
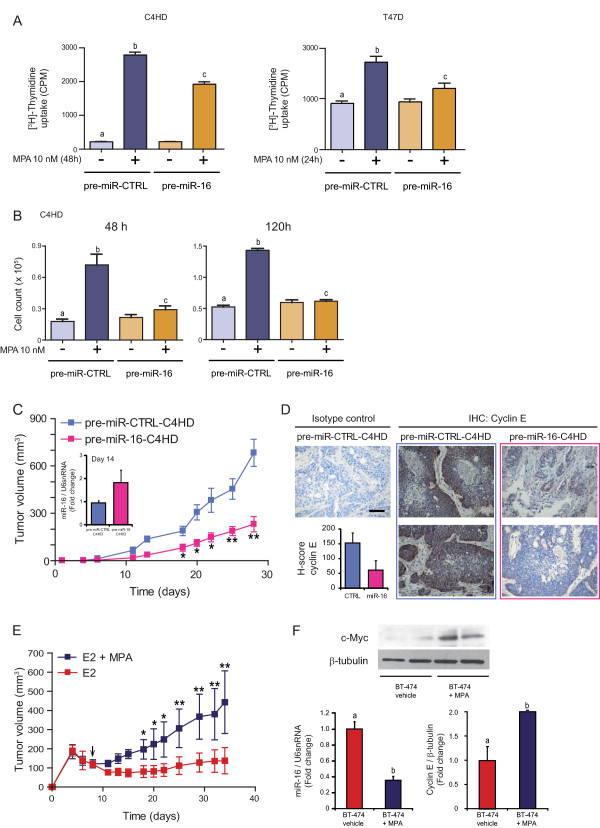
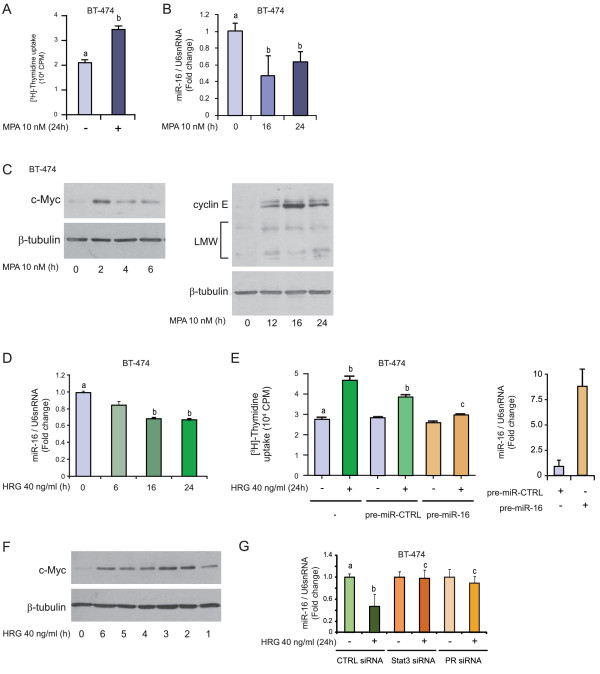
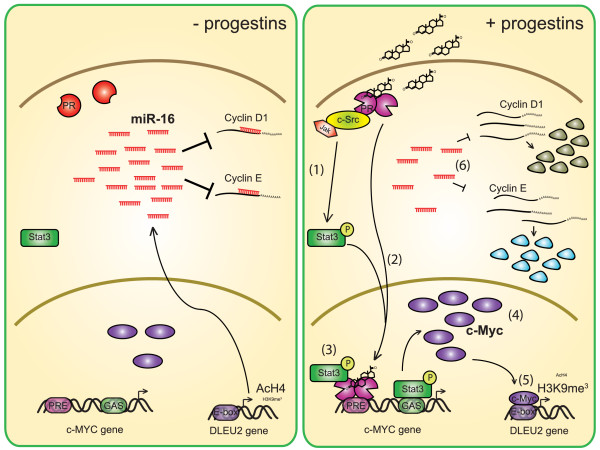
Results: We found that nine miRNAs were upregulated and seven were downregulated by progestin in mammary tumor cells. miR-16, whose function as a tumor suppressor in leukemia has already been shown, was identified as one of the downregulated miRNAs in murine and human breast cancer cells. Progestin induced a decrease in miR-16 levels via the classical PR and through a hierarchical interplay between Stat3 and the oncogenic transcription factor c-Myc. A search for miR-16 targets showed that the CCNE1 gene, encoding the cell cycle regulator cyclin E, contains conserved putative miR-16 target sites in its mRNA 3' UTR region. We found that, similar to the molecular mechanism underlying progestin-modulated miR-16 expression, Stat3 and c-Myc participated in the induction of cyclin E expression by progestin. Moreover, overexpression of miR-16 abrogated the ability of progestin to induce cyclin E upregulation, revealing that cyclin E is a novel target of miR-16 in breast cancer. Overexpression of miR-16 also inhibited progestin-induced breast tumor growth in vitro and in vivo, demonstrating for the first time, a role for miR-16 as a tumor suppressor in mammary tumorigenesis. We also found that the ErbB ligand heregulin (HRG) downregulated the expression of miR-16, which then participates in the proliferative activity of HRG in breast tumor cells.
Conclusions: In this study, we reveal the first progestin-regulated miRNA expression profile and identify a novel role for miR-16 as a tumor suppressor in progestin- and growth factor-induced growth in breast cancer.
Figures
Similar articles
-
Progesterone receptor activation downregulates GATA3 by transcriptional repression and increased protein turnover promoting breast tumor growth.Breast Cancer Res. 2014 Dec 6;16(6):491. doi: 10.1186/s13058-014-0491-x. Breast Cancer Res. 2014. PMID: 25479686 Free PMC article.
-
MiR-137 targets estrogen-related receptor alpha and impairs the proliferative and migratory capacity of breast cancer cells.PLoS One. 2012;7(6):e39102. doi: 10.1371/journal.pone.0039102. Epub 2012 Jun 18. PLoS One. 2012. PMID: 22723937 Free PMC article.
-
MiR-15a is underexpressed and inhibits the cell cycle by targeting CCNE1 in breast cancer.Int J Oncol. 2013 Oct;43(4):1212-8. doi: 10.3892/ijo.2013.2034. Epub 2013 Jul 23. Int J Oncol. 2013. PMID: 23900351
-
From microRNA functions to microRNA therapeutics: novel targets and novel drugs in breast cancer research and treatment (Review).Int J Oncol. 2013 Oct;43(4):985-94. doi: 10.3892/ijo.2013.2059. Epub 2013 Aug 12. Int J Oncol. 2013. PMID: 23939688 Free PMC article. Review.
-
Progesterone regulation of stem and progenitor cells in normal and malignant breast.Mol Cell Endocrinol. 2012 Jun 24;357(1-2):71-9. doi: 10.1016/j.mce.2011.09.021. Epub 2011 Sep 16. Mol Cell Endocrinol. 2012. PMID: 21945473 Free PMC article. Review.
Cited by
-
Air pollution-induced placental epigenetic alterations in early life: a candidate miRNA approach.Epigenetics. 2018;13(2):135-146. doi: 10.1080/15592294.2016.1155012. Epub 2018 Mar 5. Epigenetics. 2018. PMID: 27104955 Free PMC article.
-
miR15a and miR16 in Chilean type 1 diabetes patients: possible association with apoptosis, inflammatory, or autoimmunity markers.J Endocrinol Invest. 2018 Sep;41(9):1083-1088. doi: 10.1007/s40618-018-0837-9. Epub 2018 Jan 30. J Endocrinol Invest. 2018. PMID: 29383679
-
Inhibition of miR301 enhances Akt-mediated cell proliferation by accumulation of PTEN in nucleus and its effects on cell-cycle regulatory proteins.Oncotarget. 2016 Apr 12;7(15):20953-65. doi: 10.18632/oncotarget.7996. Oncotarget. 2016. PMID: 26967567 Free PMC article.
-
The oncomiR miR-197 is a novel prognostic indicator for non-small cell lung cancer patients.Br J Cancer. 2015 Apr 28;112(9):1527-35. doi: 10.1038/bjc.2015.119. Epub 2015 Mar 31. Br J Cancer. 2015. PMID: 25867273 Free PMC article.
-
Regulation of breast cancer metastasis signaling by miRNAs.Cancer Metastasis Rev. 2020 Sep;39(3):837-886. doi: 10.1007/s10555-020-09905-7. Cancer Metastasis Rev. 2020. PMID: 32577859 Free PMC article. Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
