

Estimating RNA-quality using GeneChip microarrays
- PMID: 22583818
- PMCID: PMC3519671
- DOI: 10.1186/1471-2164-13-186
Estimating RNA-quality using GeneChip microarrays
Abstract
Background: Microarrays are a powerful tool for transcriptome analysis. Best results are obtained using high-quality RNA samples for preparation and hybridization. Issues with RNA integrity can lead to low data quality and failure of the microarray experiment.
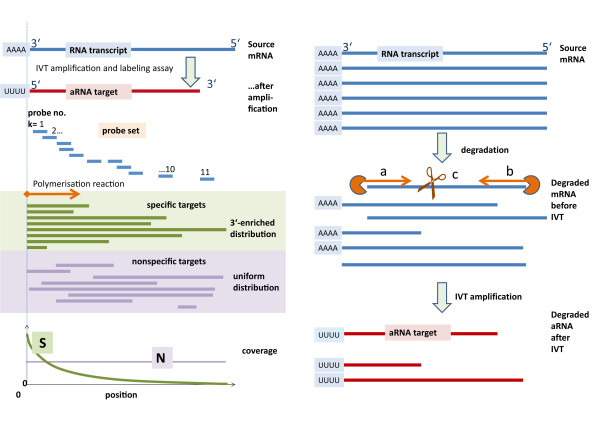
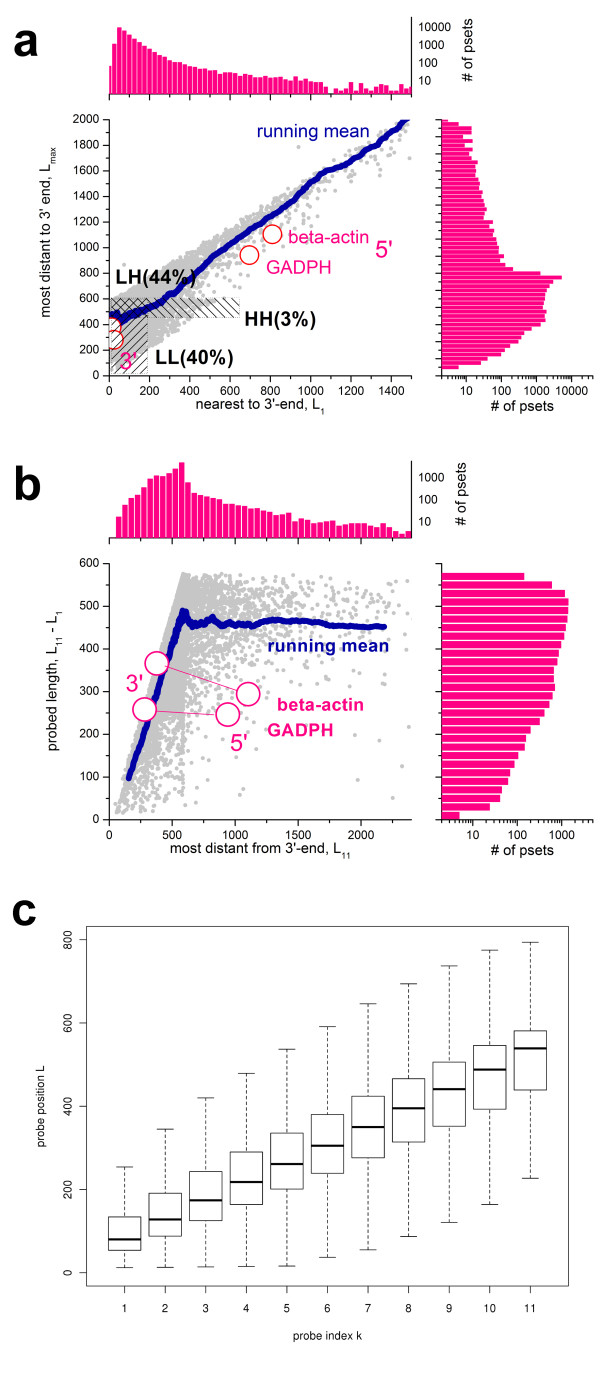
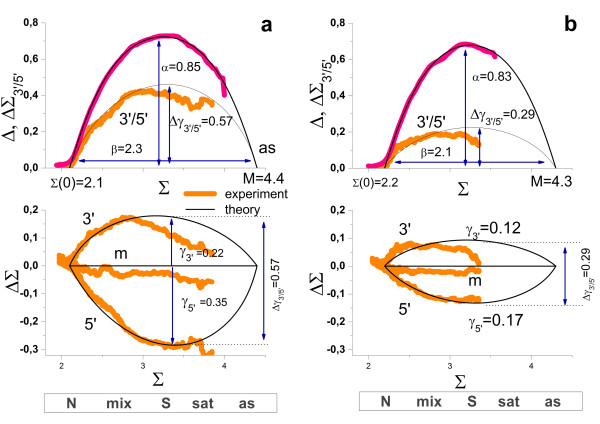
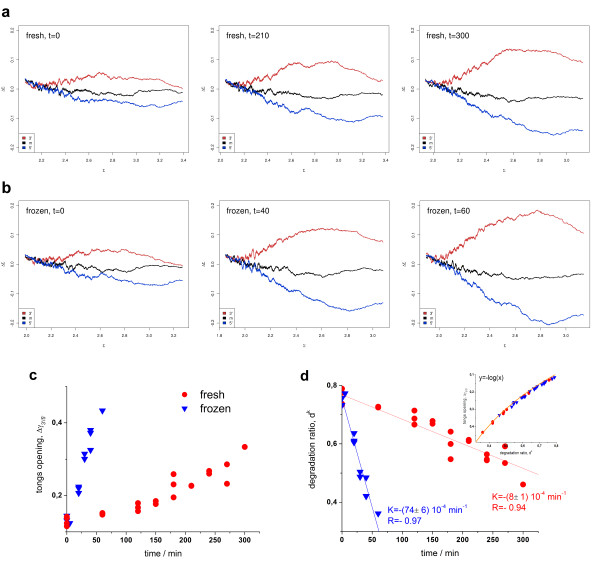
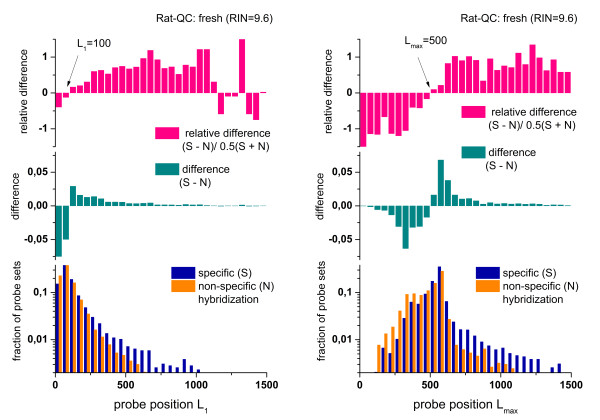
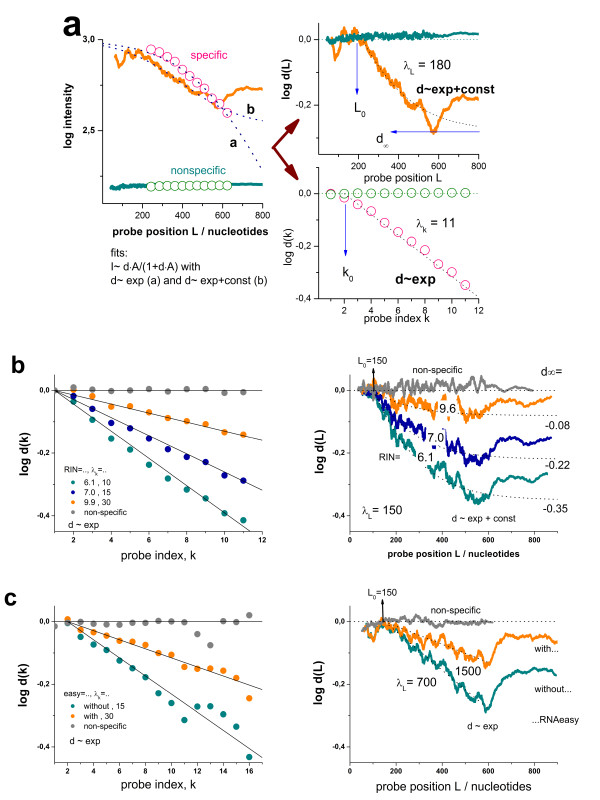
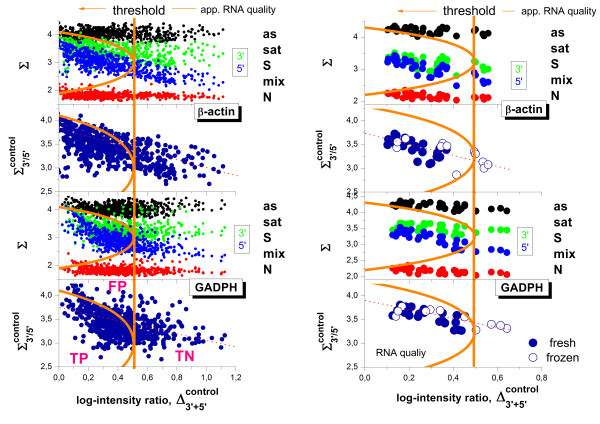
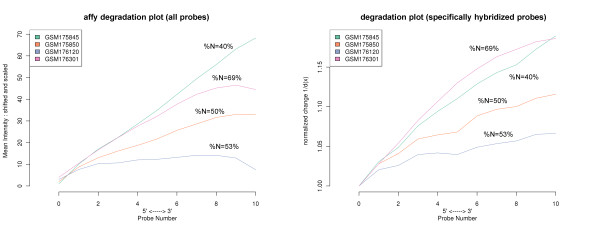
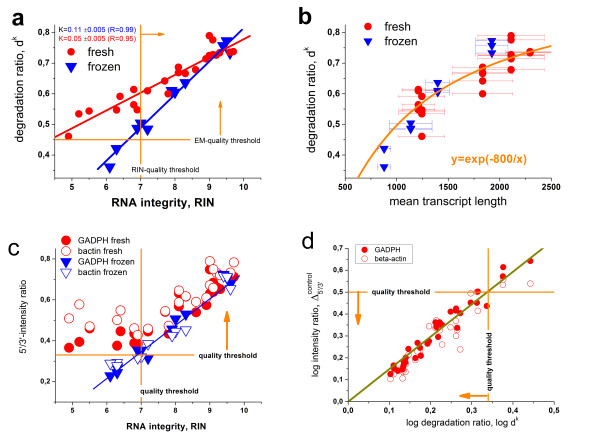
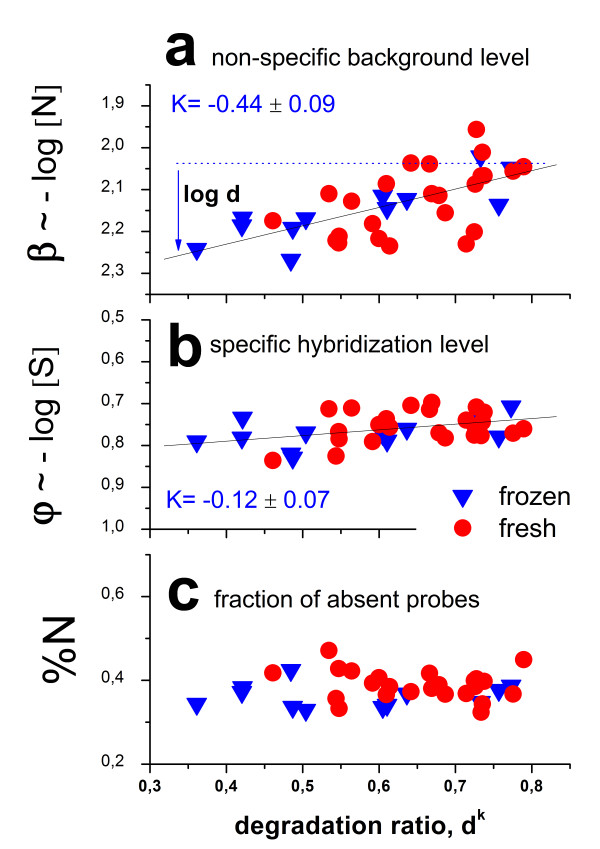
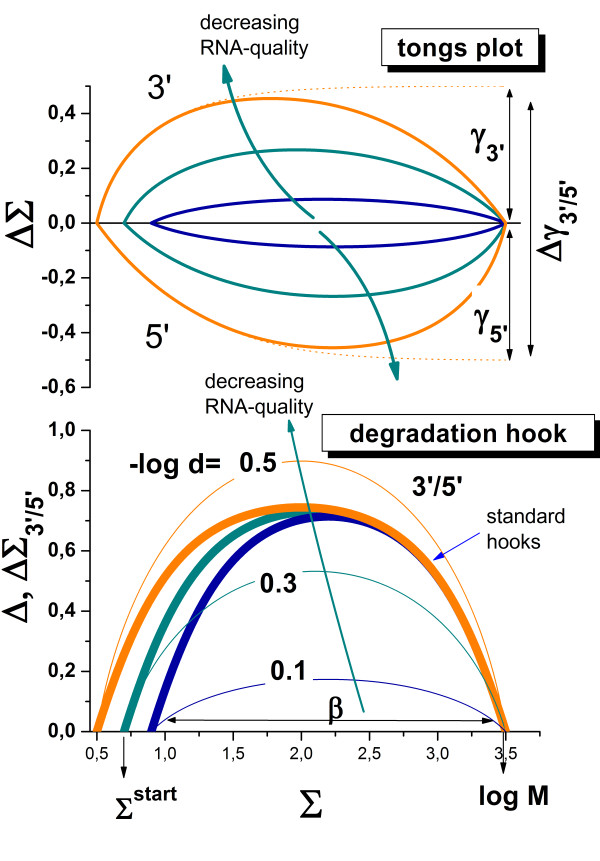
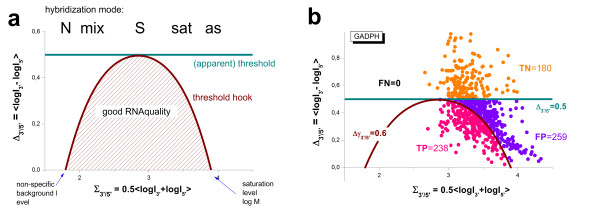
Results: Microarray intensity data contains information to estimate the RNA quality of the sample. We here study the interplay of the characteristics of RNA surface hybridization with the effects of partly truncated transcripts on probe intensity. The 3'/5' intensity gradient, the basis of microarray RNA quality measures, is shown to depend on the degree of competitive binding of specific and of non-specific targets to a particular probe, on the degree of saturation of the probes with bound transcripts and on the distance of the probe from the 3'-end of the transcript. Increasing degrees of non-specific hybridization or of saturation reduce the 3'/5' intensity gradient and if not taken into account, this leads to biased results in common quality measures for GeneChip arrays such as affyslope or the control probe intensity ratio. We also found that short probe sets near the 3'-end of the transcripts are prone to non-specific hybridization presumable because of inaccurate positional assignment and the existence of transcript isoforms with variable 3' UTRs. Poor RNA quality is associated with a decreased amount of RNA material hybridized on the array paralleled by a decreased total signal level. Additionally, it causes a gene-specific loss of signal due to the positional bias of transcript abundance which requires an individual, gene-specific correction. We propose a new RNA quality measure that considers the hybridization mode. Graphical characteristics are introduced allowing assessment of RNA quality of each single array ('tongs plot' and 'degradation hook'). Furthermore, we suggest a method to correct for effects of RNA degradation on microarray intensities.
Conclusions: The presented RNA degradation measure has best correlation with the independent RNA integrity measure RIN, and therefore presents itself as a valuable tool for quality control and even for the study of RNA degradation. When RNA degradation effects are detected in microarray experiments, a correction of the induced bias in probe intensities is advised.
Figures
Similar articles
-
AffyRNADegradation: control and correction of RNA quality effects in GeneChip expression data.Bioinformatics. 2013 Jan 1;29(1):129-31. doi: 10.1093/bioinformatics/bts629. Epub 2012 Oct 24. Bioinformatics. 2013. PMID: 23097420 Free PMC article.
-
Base pair interactions and hybridization isotherms of matched and mismatched oligonucleotide probes on microarrays.Langmuir. 2005 Sep 27;21(20):9287-302. doi: 10.1021/la051231s. Langmuir. 2005. PMID: 16171364
-
Quality assessment and data handling methods for Affymetrix Gene 1.0 ST arrays with variable RNA integrity.BMC Genomics. 2013 Jan 16;14:14. doi: 10.1186/1471-2164-14-14. BMC Genomics. 2013. PMID: 23324084 Free PMC article.
-
Effect of RNA quality on transcript intensity levels in microarray analysis of human post-mortem brain tissues.BMC Genomics. 2008 Feb 25;9:91. doi: 10.1186/1471-2164-9-91. BMC Genomics. 2008. PMID: 18298816 Free PMC article.
-
Reliability and reproducibility issues in DNA microarray measurements.Trends Genet. 2006 Feb;22(2):101-9. doi: 10.1016/j.tig.2005.12.005. Epub 2005 Dec 27. Trends Genet. 2006. PMID: 16380191 Free PMC article. Review.
Cited by
-
Immunological landscape of consensus clusters in colorectal cancer.Oncotarget. 2017 Oct 27;8(62):105299-105311. doi: 10.18632/oncotarget.22169. eCollection 2017 Dec 1. Oncotarget. 2017. PMID: 29285252 Free PMC article.
-
Characterization of RNA isolated from eighteen different human tissues: results from a rapid human autopsy program.Cell Tissue Bank. 2016 Sep;17(3):361-75. doi: 10.1007/s10561-016-9555-8. Epub 2016 Apr 16. Cell Tissue Bank. 2016. PMID: 27083469 Free PMC article.
-
Whole-transcriptome, high-throughput RNA sequence analysis of the bovine macrophage response to Mycobacterium bovis infection in vitro.BMC Genomics. 2013 Apr 8;14:230. doi: 10.1186/1471-2164-14-230. BMC Genomics. 2013. PMID: 23565803 Free PMC article.
-
Cryo-Gel embedding compound for renal biopsy biobanking.Sci Rep. 2019 Oct 24;9(1):15250. doi: 10.1038/s41598-019-51962-8. Sci Rep. 2019. PMID: 31649317 Free PMC article.
-
AffyRNADegradation: control and correction of RNA quality effects in GeneChip expression data.Bioinformatics. 2013 Jan 1;29(1):129-31. doi: 10.1093/bioinformatics/bts629. Epub 2012 Oct 24. Bioinformatics. 2013. PMID: 23097420 Free PMC article.
References
-
- Lee J, Hever A, Willhite D, Zlotnik A, Hevezi P. Effects of RNA degradation on gene expression analysis of human postmortem tissues. FASEB J. 2005;04:3552fje. - PubMed
-
- Copois V, Bibeau F, Bascoul-Mollevi C, Salvetat N, Chalbos P, Bareil C, Candeil L, Fraslon C, Conseiller E, Granci V, Mazière P, Kramar A, Ychou M, Pau B, Martineau P, Molina F, Rio MD. Impact of RNA degradation on gene expression profiles: Assessment of different methods to reliably determine RNA quality. J Biotechnol. 2007;127:549–559. doi: 10.1016/j.jbiotec.2006.07.032. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
