A new strategy to reduce allelic bias in RNA-Seq readmapping
- PMID: 22584625
- PMCID: PMC3439884
- DOI: 10.1093/nar/gks425
A new strategy to reduce allelic bias in RNA-Seq readmapping
Abstract
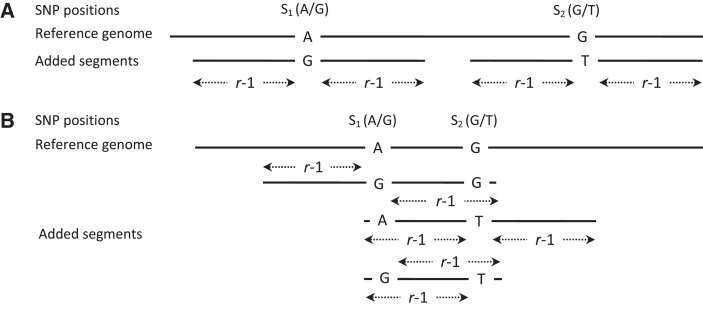
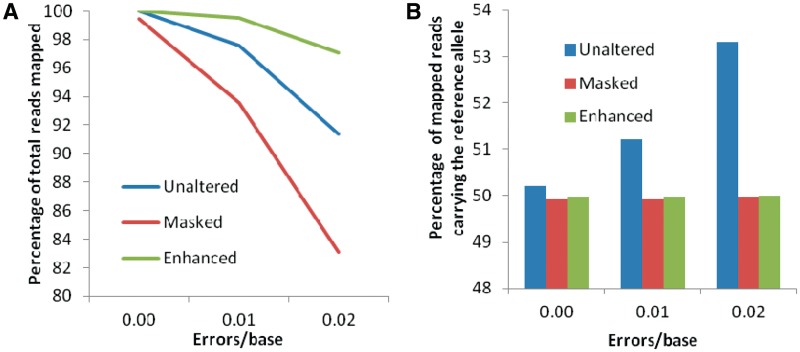
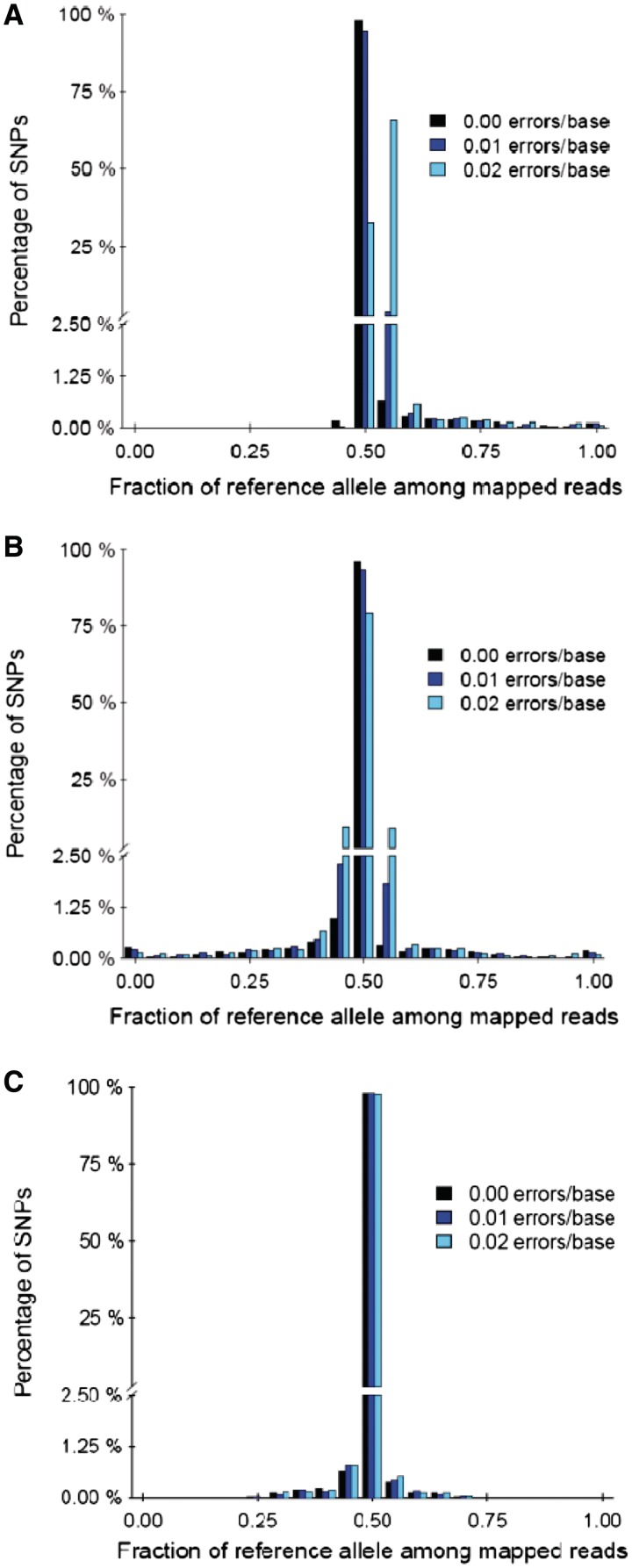
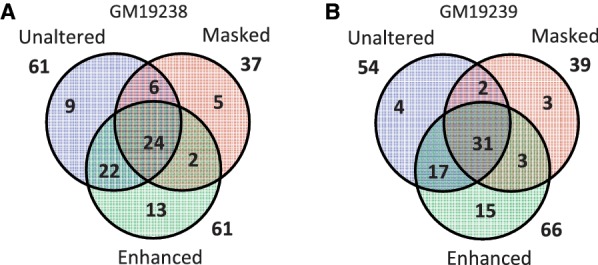
Accurate estimation of expression levels from RNA-Seq data entails precise mapping of the sequence reads to a reference genome. Because the standard reference genome contains only one allele at any given locus, reads overlapping polymorphic loci that carry a non-reference allele are at least one mismatch away from the reference and, hence, are less likely to be mapped. This bias in read mapping leads to inaccurate estimates of allele-specific expression (ASE). To address this read-mapping bias, we propose the construction of an enhanced reference genome that includes the alternative alleles at known polymorphic loci. We show that mapping to this enhanced reference reduced the read-mapping biases, leading to more reliable estimates of ASE. Experiments on simulated data show that the proposed strategy reduced the number of loci with mapping bias by ≥ 63% when compared with a previous approach that relies on masking the polymorphic loci and by ≥ 18% when compared with the standard approach that uses an unaltered reference. When we applied our strategy to actual RNA-Seq data, we found that it mapped up to 15% more reads than the previous approaches and identified many seemingly incorrect inferences made by them.
Figures