

The OPCML tumor suppressor functions as a cell surface repressor-adaptor, negatively regulating receptor tyrosine kinases in epithelial ovarian cancer
- PMID: 22585860
- PMCID: PMC3378039
- DOI: 10.1158/2159-8290.CD-11-0256
The OPCML tumor suppressor functions as a cell surface repressor-adaptor, negatively regulating receptor tyrosine kinases in epithelial ovarian cancer
Abstract
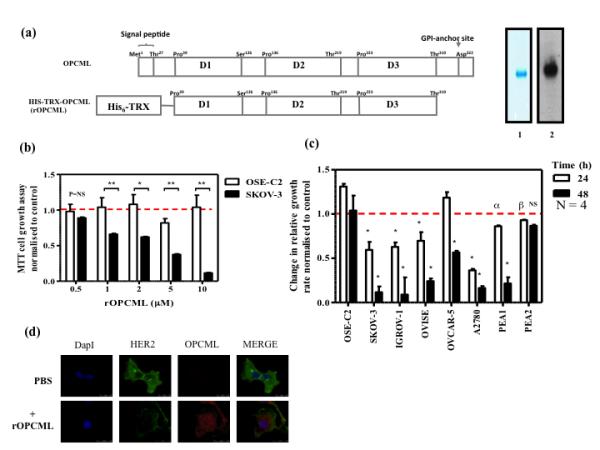
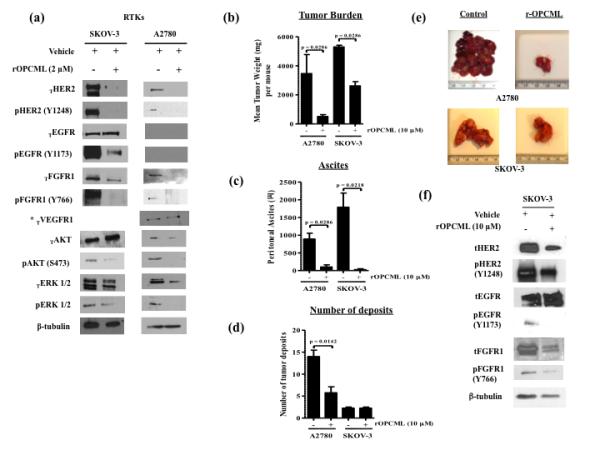
Epithelial ovarian cancer is the leading cause of death from gynecologic malignancy, and its molecular basis is poorly understood. We previously demonstrated that opioid binding protein cell adhesion molecule (OPCML) was frequently epigenetically inactivated in epithelial ovarian cancers, with tumor suppressor function in vitro and in vivo. Here, we further show the clinical relevance of OPCML and demonstrate that OPCML functions by a novel mechanism in epithelial ovarian cancer cell lines and normal ovarian surface epithelial cells by regulating a specific repertoire of receptor tyrosine kinases: EPHA2, FGFR1, FGFR3, HER2, and HER4. OPCML negatively regulates receptor tyrosine kinases by binding their extracellular domains, altering trafficking via nonclathrin-dependent endocytosis, and promoting their degradation via a polyubiquitination-associated proteasomal mechanism leading to signaling and growth inhibition. Exogenous recombinant OPCML domain 1-3 protein inhibited the cell growth of epithelial ovarian cancers cell in vitro and in vivo in 2 murine ovarian cancer intraperitoneal models that used an identical mechanism. These findings demonstrate a novel mechanism of OPCML-mediated tumor suppression and provide a proof-of-concept for recombinant OPCML protein therapy in epithelial ovarian cancers.
Significance: The OPCML tumor suppressor negatively regulates a specific spectrum of receptor tyrosine kinases in ovarian cancer cells by binding to their extracellular domain and altering trafficking to a nonclathrin, caveolin-1–associated endosomal pathway that results in receptor tyrosine kinase polyubiquitination and proteasomal degradation. Recombinant OPCML domain 1-3 recapitulates this mechanism and may allow for the implementation of an extracellular tumor-suppressor replacement strategy.
©2012 AACR.
Figures

Comment in
-
New roles opined for OPCML.Cancer Discov. 2012 Feb;2(2):115-6. doi: 10.1158/2159-8290.CD-11-0356. Cancer Discov. 2012. PMID: 22585855 Free PMC article.
References
-
- Statistics UKofN . Cancer trends in England and Wales 1950-1999. Office of NationalStatistics; 2002.
-
- Hall J, Paul J, Brown R. Critical evaluation of p53 as a prognostic marker in ovarian cancer. Expert Reviews in Molecular Medicine. 2004;6:1–20. - PubMed
-
- Radice P. Mutations of BRCA genes in hereditary breast and ovarian cancer. Journal of Experimental & Clinical Cancer Research. 2002;21:9–12. - PubMed
-
- Meng Q, Xia C, Fang J, Rojanasakul Y, Jiang B. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cellular Signalling. 2006;18:2262–71. - PubMed
-
- Maihle NJ, Baron AT, Barrette BA, Boardman CH, Christensen TA, Cora EM, et al. EGF/ErbB receptor family in ovarian cancer. Cancer Treat Res. 2002;107:247–58. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
