

Copy number variation detection and genotyping from exome sequence data
- PMID: 22585873
- PMCID: PMC3409265
- DOI: 10.1101/gr.138115.112
Copy number variation detection and genotyping from exome sequence data
Abstract
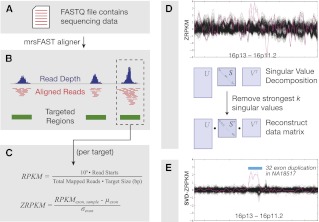
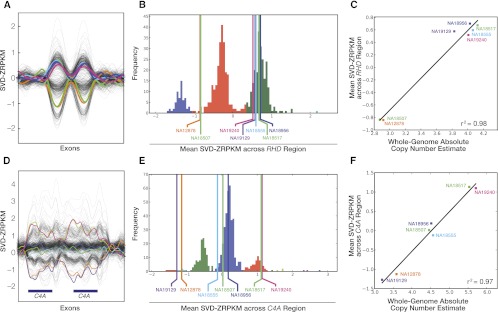
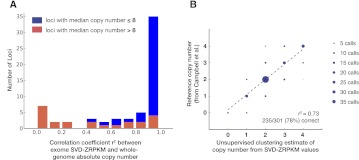
While exome sequencing is readily amenable to single-nucleotide variant discovery, the sparse and nonuniform nature of the exome capture reaction has hindered exome-based detection and characterization of genic copy number variation. We developed a novel method using singular value decomposition (SVD) normalization to discover rare genic copy number variants (CNVs) as well as genotype copy number polymorphic (CNP) loci with high sensitivity and specificity from exome sequencing data. We estimate the precision of our algorithm using 122 trios (366 exomes) and show that this method can be used to reliably predict (94% overall precision) both de novo and inherited rare CNVs involving three or more consecutive exons. We demonstrate that exome-based genotyping of CNPs strongly correlates with whole-genome data (median r(2) = 0.91), especially for loci with fewer than eight copies, and can estimate the absolute copy number of multi-allelic genes with high accuracy (78% call level). The resulting user-friendly computational pipeline, CoNIFER (copy number inference from exome reads), can reliably be used to discover disruptive genic CNVs missed by standard approaches and should have broad application in human genetic studies of disease.
Figures
References
-
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745–755 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous