

Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study
- PMID: 22585969
- PMCID: PMC3362521
- DOI: 10.1085/jgp.201210772
Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study
Abstract
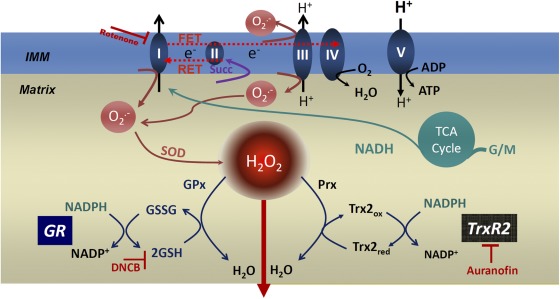
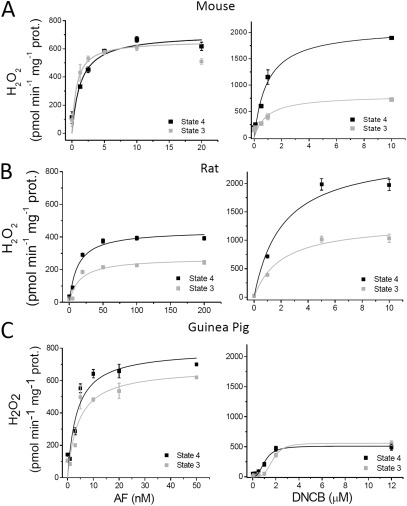
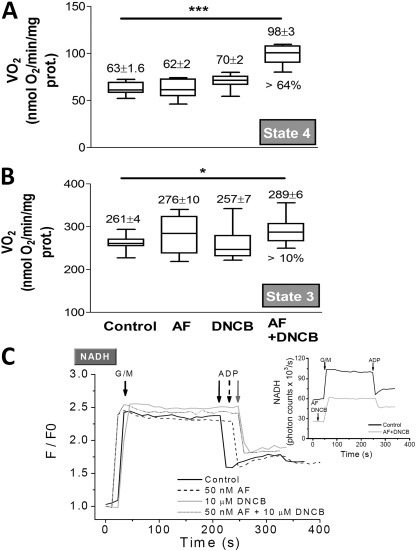
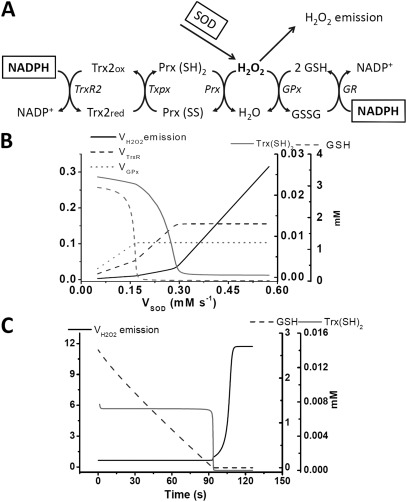
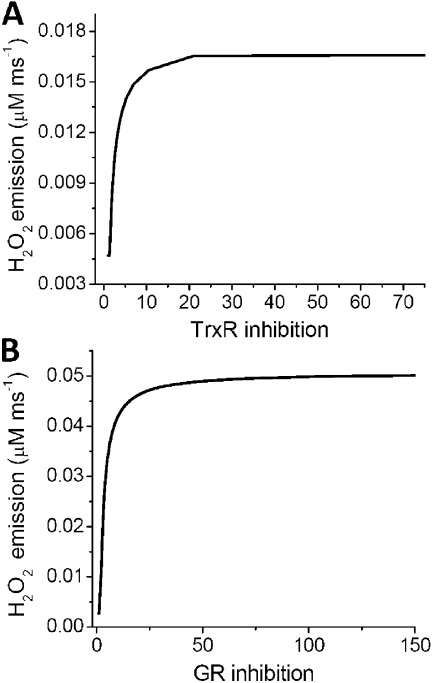
The net emission of hydrogen peroxide (H(2)O(2)) from mitochondria results from the balance between reactive oxygen species (ROS) continuously generated in the respiratory chain and ROS scavenging. The relative contribution of the two major antioxidant systems in the mitochondrial matrix, glutathione (GSH) and thioredoxin (Trx), has not been assessed. In this paper, we examine this key question via combined experimental and theoretical approaches, using isolated heart mitochondria from mouse, rat, and guinea pig. As compared with untreated control mitochondria, selective inhibition of Trx reductase with auranofin along with depletion of GSH with 2,4-dinitrochlorobenzene led to a species-dependent increase in H(2)O(2) emission flux of 17, 11, and 6 fold in state 4 and 15, 7, and 8 fold in state 3 for mouse, rat, and guinea pig mitochondria, respectively. The maximal H(2)O(2) emission as a percentage of the total O(2) consumption flux was 11%/2.3% for mouse in states 4 and 3 followed by 2%/0.25% and 0.74%/0.29% in the rat and guinea pig, respectively. A minimal computational model accounting for the kinetics of GSH/Trx systems was developed and was able to simulate increase in H(2)O(2) emission fluxes when both scavenging systems were inhibited separately or together. Model simulations suggest that GSH/Trx systems act in concert. When the scavenging capacity of either one of them saturates during H(2)O(2) overload, they relieve each other until complete saturation, when maximal ROS emission occurs. Quantitatively, these results converge on the idea that GSH/Trx scavenging systems in mitochondria are both essential for keeping minimal levels of H(2)O(2) emission, especially during state 3 respiration, when the energetic output is maximal. This suggests that the very low levels of H(2)O(2) emission observed during forward electron transport in the respiratory chain are a result of the well-orchestrated actions of the two antioxidant systems working continuously to offset ROS production.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
