

Kinetics of inhibitor cycling underlie therapeutic disparities between EGFR-driven lung and brain cancers
- PMID: 22588882
- PMCID: PMC3354705
- DOI: 10.1158/2159-8290.CD-11-0287
Kinetics of inhibitor cycling underlie therapeutic disparities between EGFR-driven lung and brain cancers
Abstract
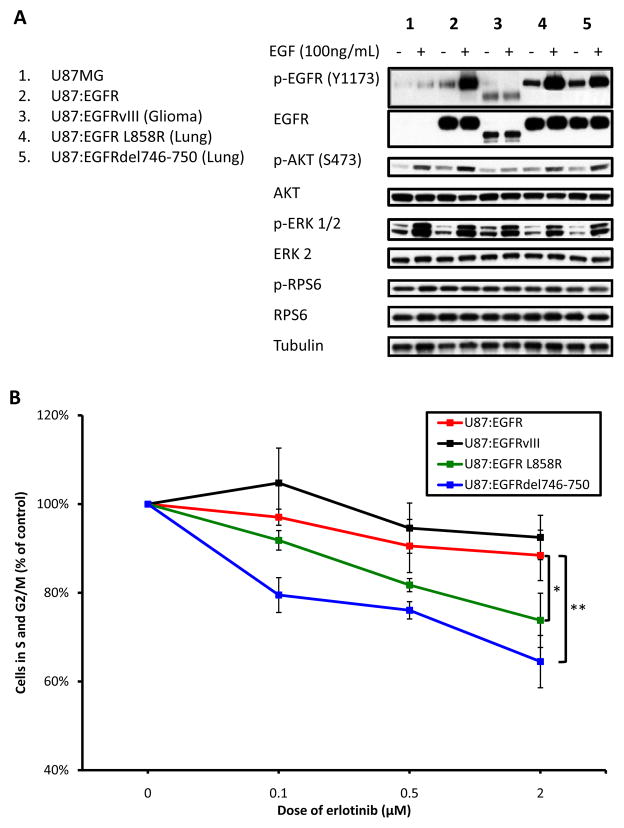
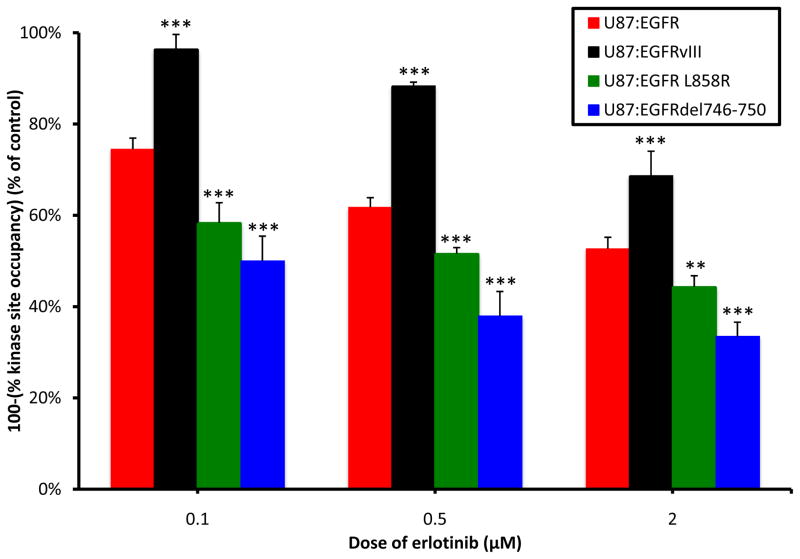
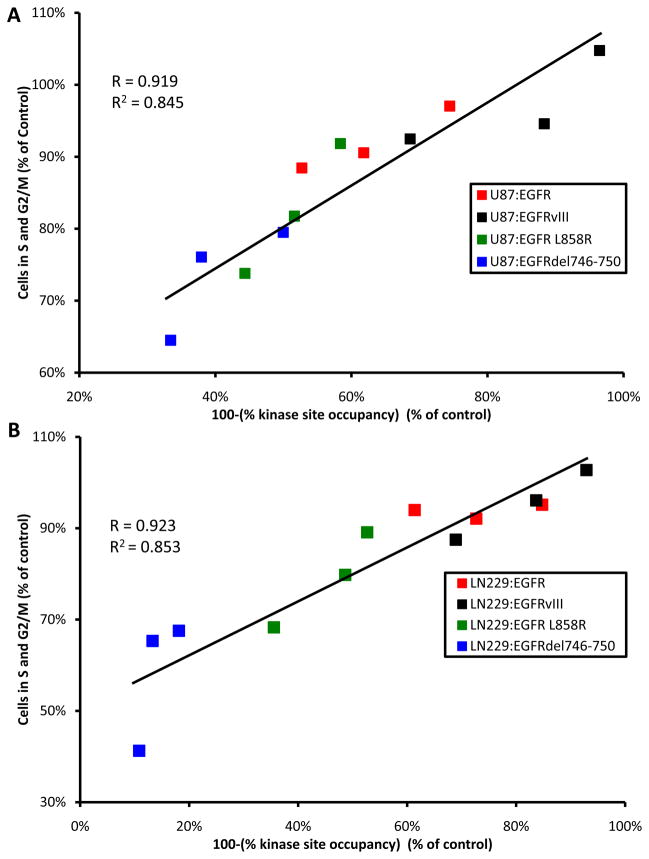
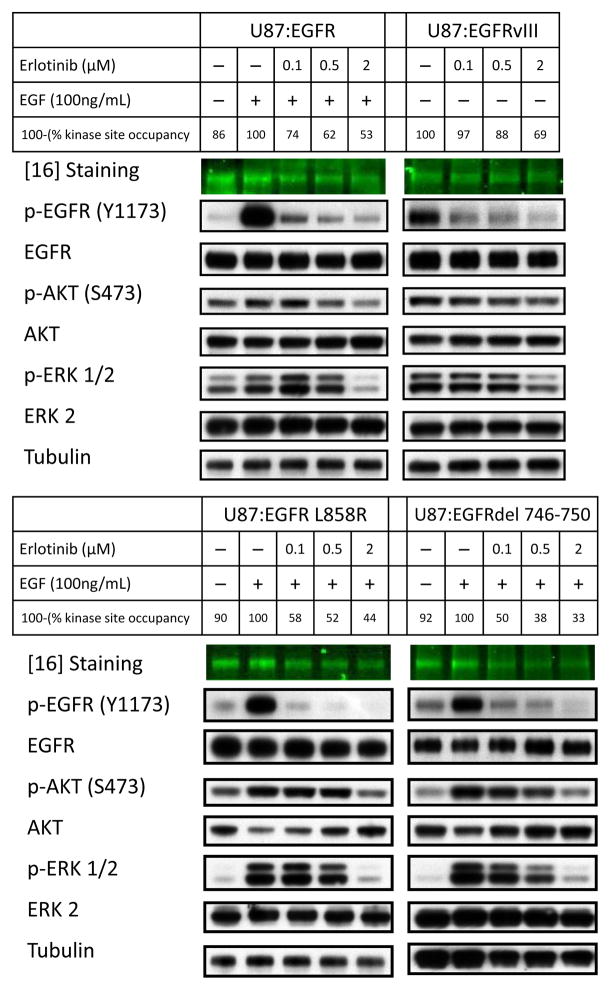
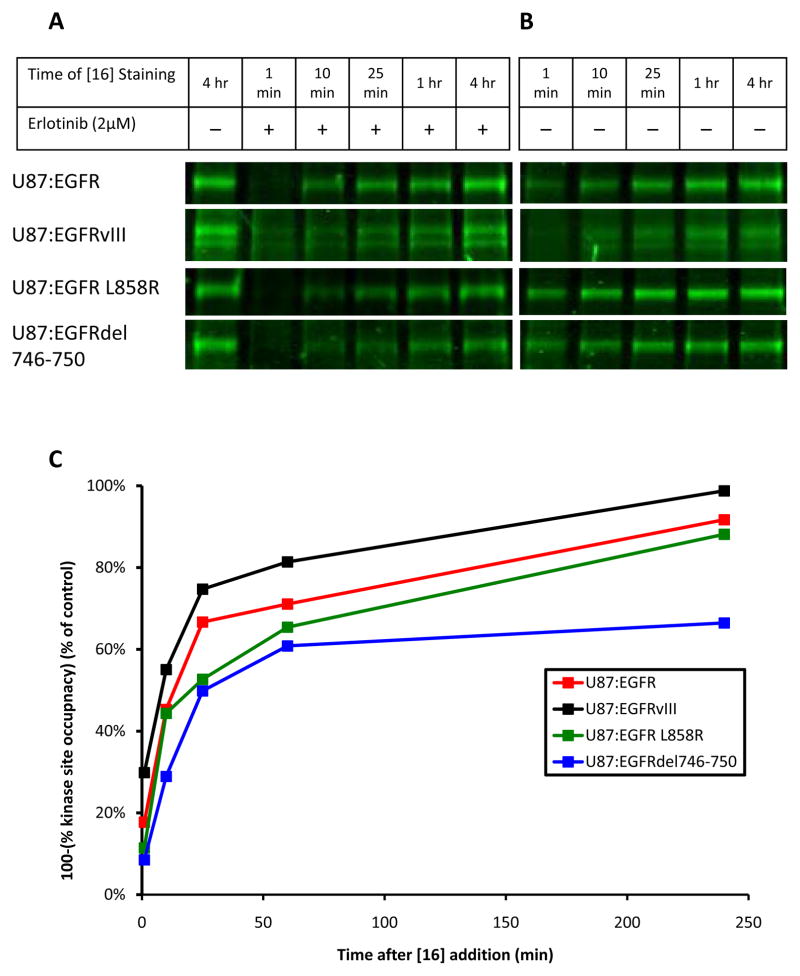
Although mutational activation of the epidermal growth factor receptor (EGFR) features prominently in glioma and non-small cell lung cancer (NSCLC), inhibitors of EGFR improve survival only in patients with NCSLC. To understand how mutations in EGFR influence response to therapy, we generated glioma cells expressing either glioma- or NSCLC-derived alleles and quantified kinase-site occupancy by clinical inhibitors with the use of a novel affinity probe and kinetic methodology. At equivalent doses, erlotinib achieved lower kinase-site occupancy in glioma-derived EGFRvIII compared with NSCLC-derived EGFR mutants. Kinase-site occupancy correlated directly with cell-cycle arrest. EGFRvIII released erlotinib rapidly compared with wild-type EGFR, whereas NSCLC-derived mutants released erlotinib slowly.
Significance: These data suggest that kinase-site occupancy is a biomarker for efficacy of EGFR inhibitors, that rapid binding and release of erlotinib in glioma-derived EGFRvIII opposes the blockade of downstream signaling, and that slower cycling of erlotinib within the active site of NSCLC-derived mutants underlies their improved clinical response.
© 2012 AACR
Conflict of interest statement
We have no potential conflicts of interest to disclose.
Figures
Comment in
-
Occupy EGFR.Cancer Discov. 2012 May;2(5):398-400. doi: 10.1158/2159-8290.CD-12-0144. Cancer Discov. 2012. PMID: 22588876 Free PMC article.
References
-
- Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res. 2003;284:99–110. - PubMed
-
- Zandi R, Larsen AB, Andersen P, Stockhausen M-R, Poulsen HS. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal. 2007;19:2013–23. - PubMed
-
- Frederick L, Wang X-Y, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–7. - PubMed
-
- Huang H-JS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji X-D, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Bio Chem. 1997;272:2927–35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
