

Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors
- PMID: 22588883
- PMCID: PMC3354723
- DOI: 10.1158/2159-8290.CD-11-0284
Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors
Abstract
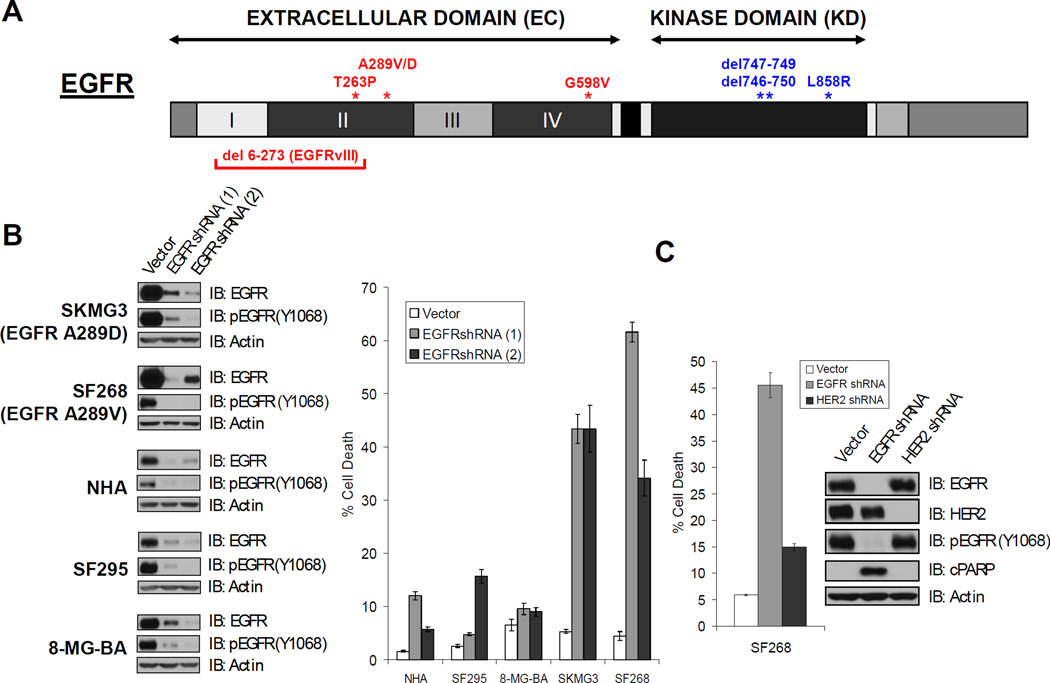
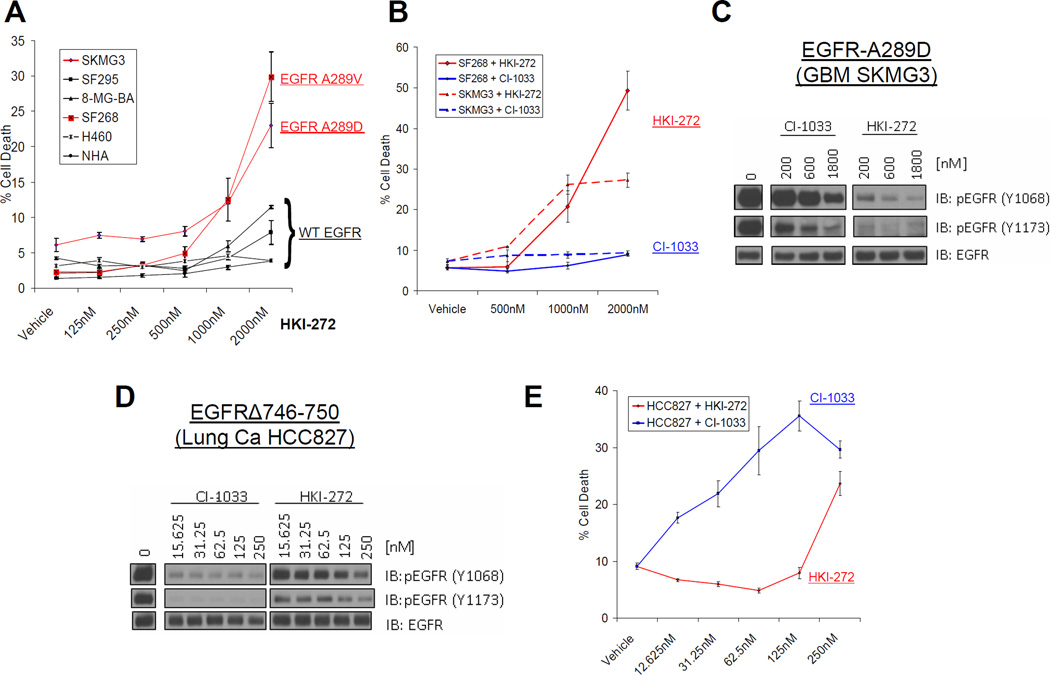
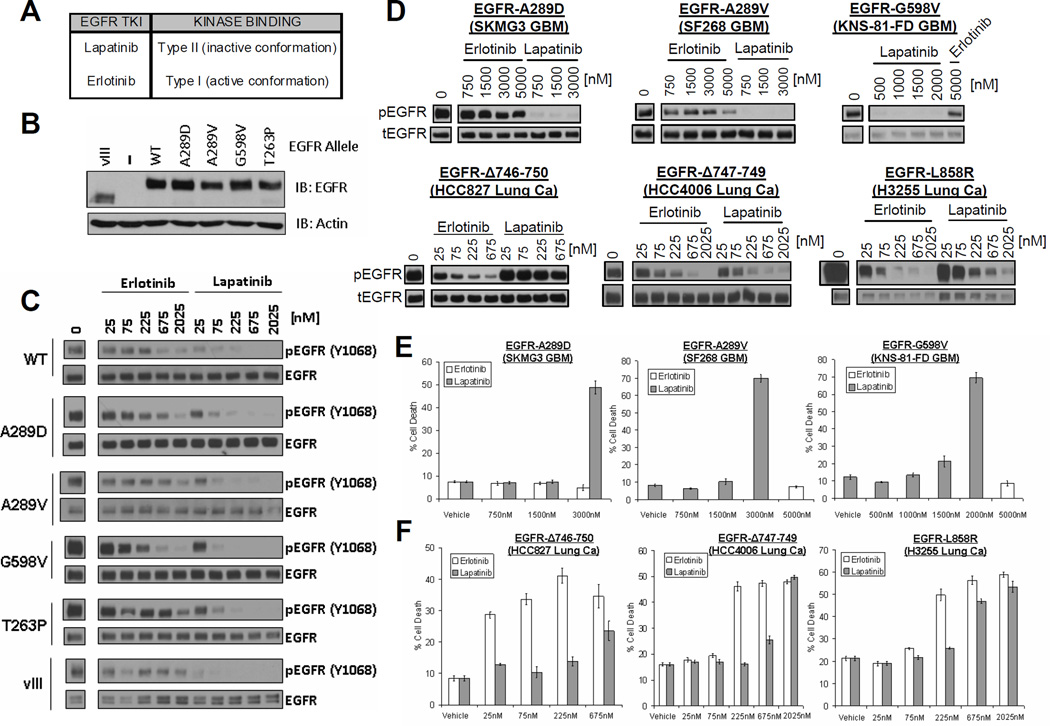
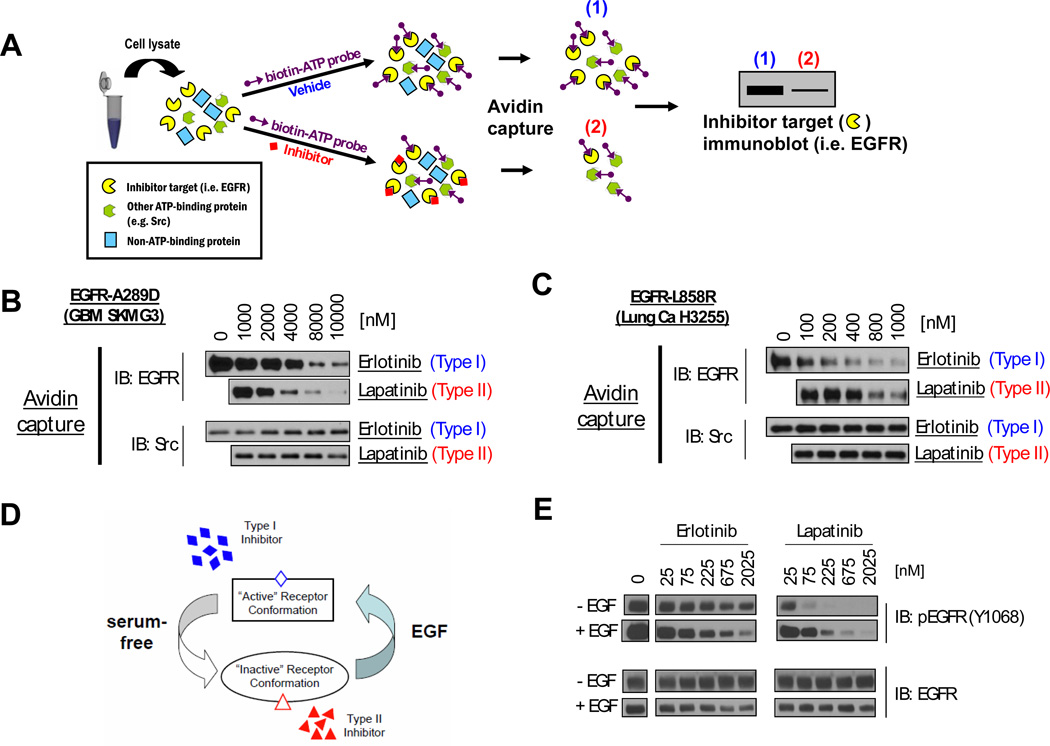
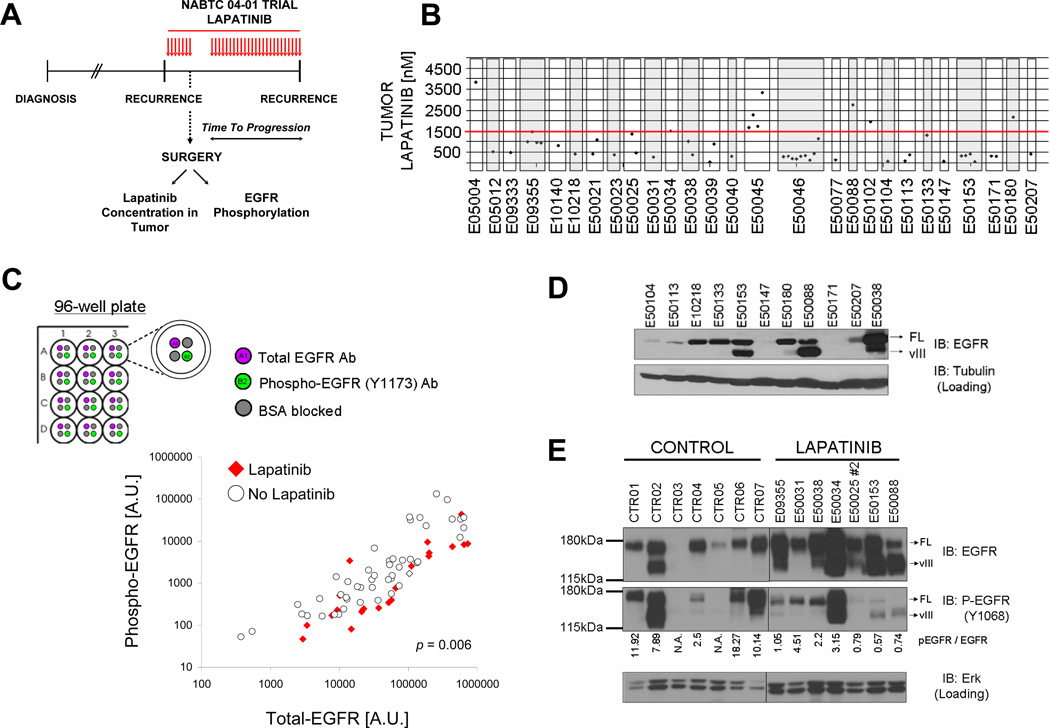
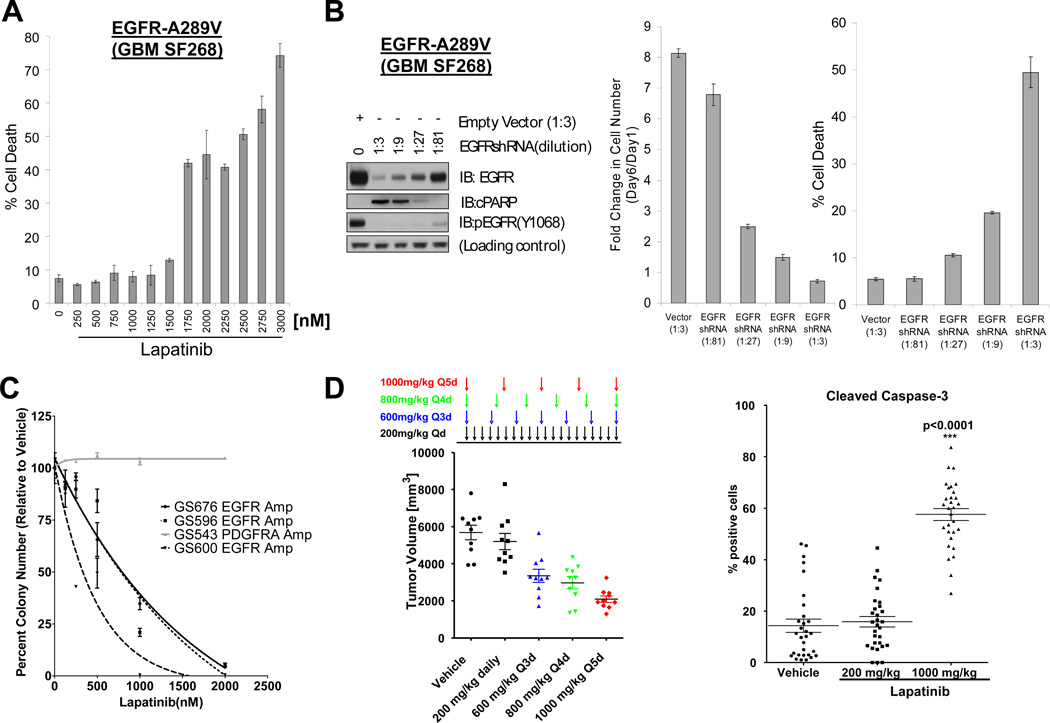
Activation of the epidermal growth factor receptor (EGFR) in glioblastoma (GBM) occurs through mutations or deletions in the extracellular (EC) domain. Unlike lung cancers with EGFR kinase domain (KD) mutations, GBMs respond poorly to the EGFR inhibitor erlotinib. Using RNAi, we show that GBM cells carrying EGFR EC mutations display EGFR addiction. In contrast to KD mutants found in lung cancer, glioma-specific EGFR EC mutants are poorly inhibited by EGFR inhibitors that target the active kinase conformation (e.g., erlotinib). Inhibitors that bind to the inactive EGFR conformation, however, potently inhibit EGFR EC mutants and induce cell death in EGFR-mutant GBM cells. Our results provide first evidence for single kinase addiction in GBM and suggest that the disappointing clinical activity of first-generation EGFR inhibitors in GBM versus lung cancer may be attributed to the different conformational requirements of mutant EGFR in these 2 cancer types.
Significance: Approximately 40% of human glioblastomas harbor oncogenic EGFR alterations, but attempts to therapeutically target EGFR with first-generation EGFR kinase inhibitors have failed. Here, we demonstrate selective sensitivity of glioma-specific EGFR mutants to ATP-site competitive EGFR kinase inhibitors that target the inactive conformation of the catalytic domain.
© 2012 AACR
Figures
Comment in
-
Targeted therapies: Glioma--it's all in the site occupancy.Nat Rev Clin Oncol. 2012 Apr 17;9(6):308. doi: 10.1038/nrclinonc.2012.65. Nat Rev Clin Oncol. 2012. PMID: 22508031 No abstract available.
-
Occupy EGFR.Cancer Discov. 2012 May;2(5):398-400. doi: 10.1158/2159-8290.CD-12-0144. Cancer Discov. 2012. PMID: 22588876 Free PMC article.
References
-
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008 Jul 31;359(5):492–507. - PubMed
-
- Sawyers CL. Shifting paradigms: the seeds of oncogene addiction. Nat Med. 2009 Oct;15(10):1158–1161. - PubMed
-
- Sellers WR. A blueprint for advancing genetics-based cancer therapy. Cell. 2011 Sep 30;147(1):26–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 CA141502/CA/NCI NIH HHS/United States
- UL1 RR024153/RR/NCRR NIH HHS/United States
- M01 RR003186/RR/NCRR NIH HHS/United States
- U01CA62407-08/CA/NCI NIH HHS/United States
- U01 CA062399/CA/NCI NIH HHS/United States
- CA62422/CA/NCI NIH HHS/United States
- U01 CA062421/CA/NCI NIH HHS/United States
- U01CA62405/CA/NCI NIH HHS/United States
- U01 CA062422/CA/NCI NIH HHS/United States
- CA62399/CA/NCI NIH HHS/United States
- U01 CA062407/CA/NCI NIH HHS/United States
- U01 CA062426/CA/NCI NIH HHS/United States
- M01 RR000079/RR/NCRR NIH HHS/United States
- UL1 TR000005/TR/NCATS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- M01-RR03186/RR/NCRR NIH HHS/United States
- M01-RR0865/RR/NCRR NIH HHS/United States
- U54 CA143798/CA/NCI NIH HHS/United States
- M01 RR000056/RR/NCRR NIH HHS/United States
- K08 NS062907/NS/NINDS NIH HHS/United States
- M01-RR00079/RR/NCRR NIH HHS/United States
- CA16672/CA/NCI NIH HHS/United States
- M01 RR000865/RR/NCRR NIH HHS/United States
- 5-U01CA62399-09/CA/NCI NIH HHS/United States
- UL1 TR000124/TR/NCATS NIH HHS/United States
- M01-RR00056/RR/NCRR NIH HHS/United States
- 5K08NS062907/NS/NINDS NIH HHS/United States
- U01 CA062405/CA/NCI NIH HHS/United States
- U01 CA062412/CA/NCI NIH HHS/United States
- U01CA62399/CA/NCI NIH HHS/United States
- U01CA62421-08/CA/NCI NIH HHS/United States
- CA62426/CA/NCI NIH HHS/United States
- U54CA143798/CA/NCI NIH HHS/United States
- CA62412/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
