

Observation of dually decoded regions of the human genome using ribosome profiling data
- PMID: 22593554
- PMCID: PMC3483551
- DOI: 10.1101/gr.133249.111
Observation of dually decoded regions of the human genome using ribosome profiling data
Abstract
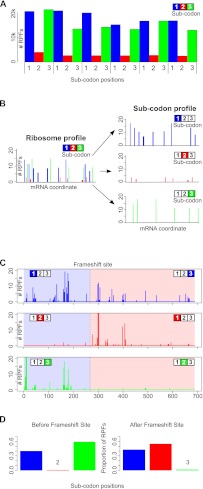
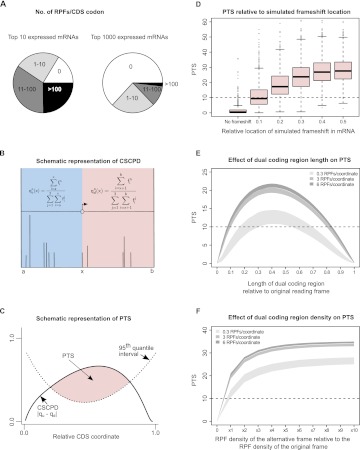
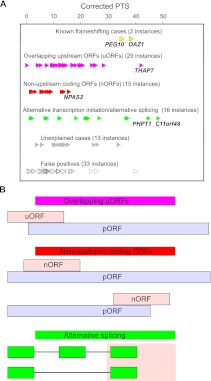
The recently developed ribosome profiling technique (Ribo-Seq) allows mapping of the locations of translating ribosomes on mRNAs with subcodon precision. When ribosome protected fragments (RPFs) are aligned to mRNA, a characteristic triplet periodicity pattern is revealed. We utilized the triplet periodicity of RPFs to develop a computational method for detecting transitions between reading frames that occur during programmed ribosomal frameshifting or in dual coding regions where the same nucleotide sequence codes for multiple proteins in different reading frames. Application of this method to ribosome profiling data obtained for human cells allowed us to detect several human genes where the same genomic segment is translated in more than one reading frame (from different transcripts as well as from the same mRNA) and revealed the translation of hitherto unpredicted coding open reading frames.
Figures



References
-
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415: 92–96 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources