

Peptide identification by tandem mass spectrometry with alternate fragmentation modes
- PMID: 22595789
- PMCID: PMC3434779
- DOI: 10.1074/mcp.R112.018556
Peptide identification by tandem mass spectrometry with alternate fragmentation modes
Abstract
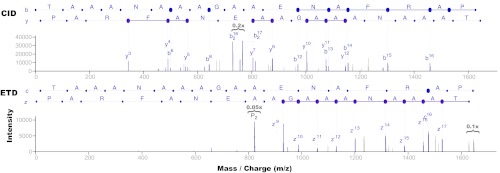
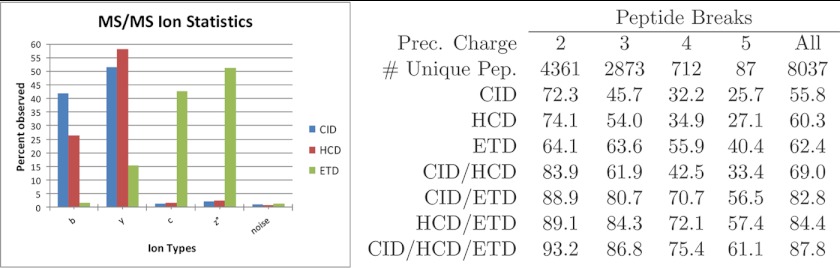
The high-throughput nature of proteomics mass spectrometry is enabled by a productive combination of data acquisition protocols and the computational tools used to interpret the resulting spectra. One of the key components in mainstream protocols is the generation of tandem mass (MS/MS) spectra by peptide fragmentation using collision induced dissociation, the approach currently used in the large majority of proteomics experiments to routinely identify hundreds to thousands of proteins from single mass spectrometry runs. Complementary to these, alternative peptide fragmentation methods such as electron capture/transfer dissociation and higher-energy collision dissociation have consistently achieved significant improvements in the identification of certain classes of peptides, proteins, and post-translational modifications. Recognizing these advantages, mass spectrometry instruments now conveniently support fine-tuned methods that automatically alternate between peptide fragmentation modes for either different types of peptides or for acquisition of multiple MS/MS spectra from each peptide. But although these developments have the potential to substantially improve peptide identification, their routine application requires corresponding adjustments to the software tools and procedures used for automated downstream processing. This review discusses the computational implications of alternative and alternate modes of MS/MS peptide fragmentation and addresses some practical aspects of using such protocols for identification of peptides and post-translational modifications.
Figures
Similar articles
-
Improving the identification rate of endogenous peptides using electron transfer dissociation and collision-induced dissociation.J Proteome Res. 2013 Dec 6;12(12):5410-21. doi: 10.1021/pr400446z. Epub 2013 Oct 3. J Proteome Res. 2013. PMID: 24032530
-
[Recent advances in glycopeptide enrichment and mass spectrometry data interpretation approaches for glycoproteomics analyses].Se Pu. 2021 Oct;39(10):1045-1054. doi: 10.3724/SP.J.1123.2021.06011. Se Pu. 2021. PMID: 34505426 Free PMC article. Review. Chinese.
-
ETISEQ--an algorithm for automated elution time ion sequencing of concurrently fragmented peptides for mass spectrometry-based proteomics.BMC Bioinformatics. 2009 Aug 10;10:244. doi: 10.1186/1471-2105-10-244. BMC Bioinformatics. 2009. PMID: 19664259 Free PMC article.
-
The spectral networks paradigm in high throughput mass spectrometry.Mol Biosyst. 2012 Oct;8(10):2535-44. doi: 10.1039/c2mb25085c. Mol Biosyst. 2012. PMID: 22610447 Free PMC article. Review.
-
Interpretation of collision-induced fragmentation tandem mass spectra of posttranslationally modified peptides.Methods Mol Biol. 2007;367:169-94. doi: 10.1385/1-59745-275-0:169. Methods Mol Biol. 2007. PMID: 17185776
Cited by
-
Active Peptide AR-9 From Eupolyphaga sinensis Reduces Blood Lipid and Hepatic Lipid Accumulation by Restoring Gut Flora and Its Metabolites in a High Fat Diet-Induced Hyperlipidemia Rat.Front Pharmacol. 2022 Sep 13;13:918505. doi: 10.3389/fphar.2022.918505. eCollection 2022. Front Pharmacol. 2022. PMID: 36176455 Free PMC article.
-
Research Progress on Mono-ADP-Ribosyltransferases in Human Cell Biology.Front Cell Dev Biol. 2022 May 16;10:864101. doi: 10.3389/fcell.2022.864101. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35652091 Free PMC article. Review.
-
PepExplorer: a similarity-driven tool for analyzing de novo sequencing results.Mol Cell Proteomics. 2014 Sep;13(9):2480-9. doi: 10.1074/mcp.M113.037002. Epub 2014 May 30. Mol Cell Proteomics. 2014. PMID: 24878498 Free PMC article.
-
Computational phosphoproteomics: from identification to localization.Proteomics. 2015 Mar;15(5-6):950-63. doi: 10.1002/pmic.201400372. Epub 2015 Feb 17. Proteomics. 2015. PMID: 25475148 Free PMC article. Review.
-
Structural studies of the human α1 glycine receptor via site-specific chemical cross-linking coupled with mass spectrometry.Biophys Rep (N Y). 2024 Dec 11;4(4):100184. doi: 10.1016/j.bpr.2024.100184. Epub 2024 Oct 10. Biophys Rep (N Y). 2024. PMID: 39393591 Free PMC article.
References
-
- Nilsson T., Mann M., Aebersold R., Yates J. R., 3rd, Bairoch A., Bergeron J. J. (2010) Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods 7, 681–685 - PubMed
-
- Larsen M. R., Trelle M. B., Thingholm T. E., Jensen O. N. (2006) Analysis of posttranslational modifications of proteins by tandem mass spectrometry. Bio. Technique, 40, 790–798 - PubMed
-
- Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 - PubMed
-
- Eng J. K., Searle B. C., Clauser K. R., Tabb D. L. (2011) A face in the crowd: recognizing peptides through database search. Mol. Cell. Proteomics 10, 10.1074/mcp.R111.009522 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
