

Whole-genome bisulfite DNA sequencing of a DNMT3B mutant patient
- PMID: 22595875
- PMCID: PMC3398983
- DOI: 10.4161/epi.20523
Whole-genome bisulfite DNA sequencing of a DNMT3B mutant patient
Abstract
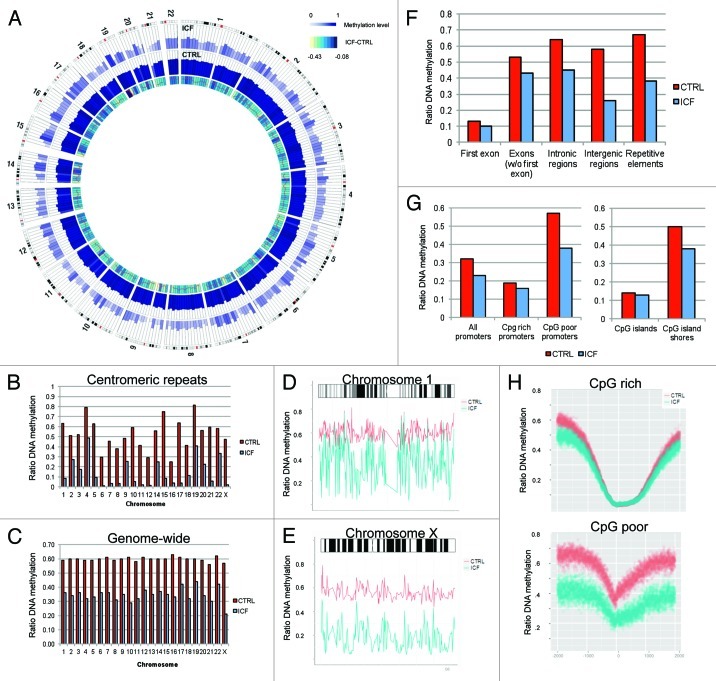
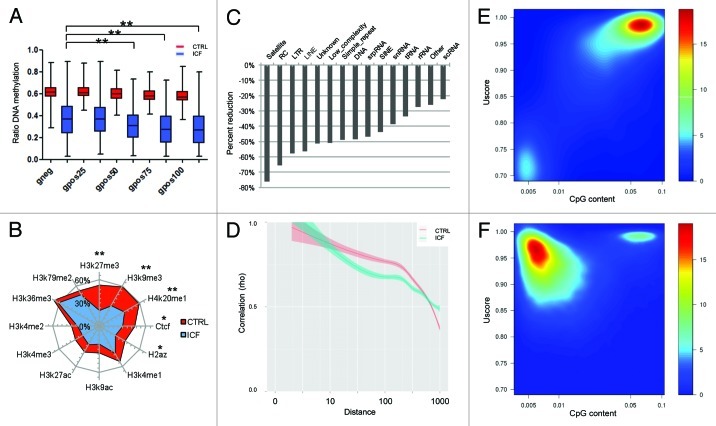
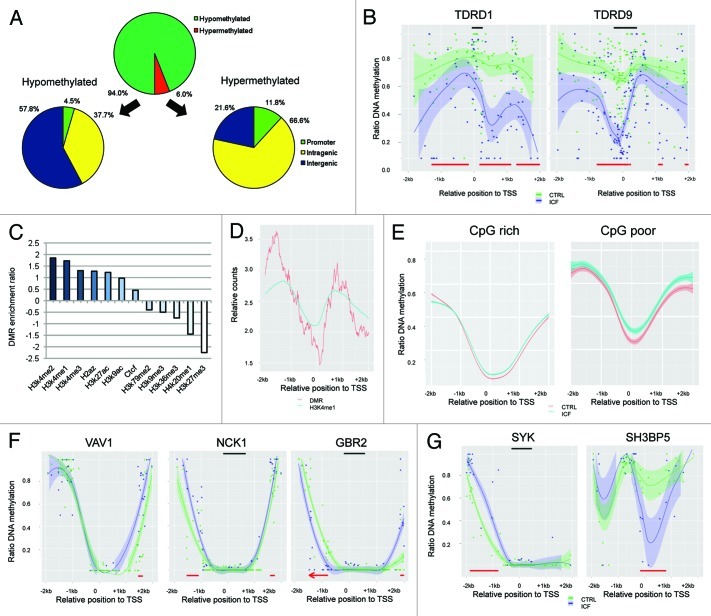
The immunodeficiency, centromere instability and facial anomalies (ICF) syndrome is associated to mutations of the DNA methyl-transferase DNMT3B, resulting in a reduction of enzyme activity. Aberrant expression of immune system genes and hypomethylation of pericentromeric regions accompanied by chromosomal instability were determined as alterations driving the disease phenotype. However, so far only technologies capable to analyze single loci were applied to determine epigenetic alterations in ICF patients. In the current study, we performed whole-genome bisulphite sequencing to assess alteration in DNA methylation at base pair resolution. Genome-wide we detected a decrease of methylation level of 42%, with the most profound changes occurring in inactive heterochromatic regions, satellite repeats and transposons. Interestingly, transcriptional active loci and ribosomal RNA repeats escaped global hypomethylation. Despite a genome-wide loss of DNA methylation the epigenetic landscape and crucial regulatory structures were conserved. Remarkably, we revealed a mislocated activity of mutant DNMT3B to H3K4me1 loci resulting in hypermethylation of active promoters. Functionally, we could associate alterations in promoter methylation with the ICF syndrome immunodeficient phenotype by detecting changes in genes related to the B-cell receptor mediated maturation pathway.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases