

Evolution and functional impact of rare coding variation from deep sequencing of human exomes
- PMID: 22604720
- PMCID: PMC3708544
- DOI: 10.1126/science.1219240
Evolution and functional impact of rare coding variation from deep sequencing of human exomes
Abstract
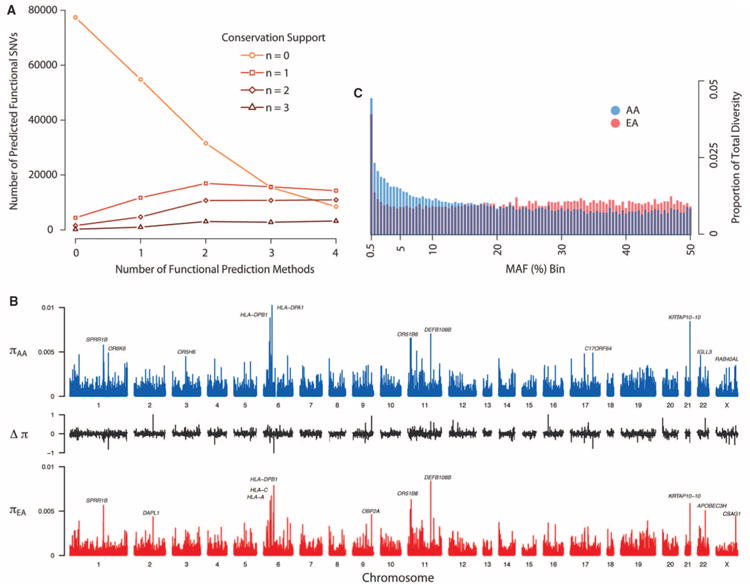
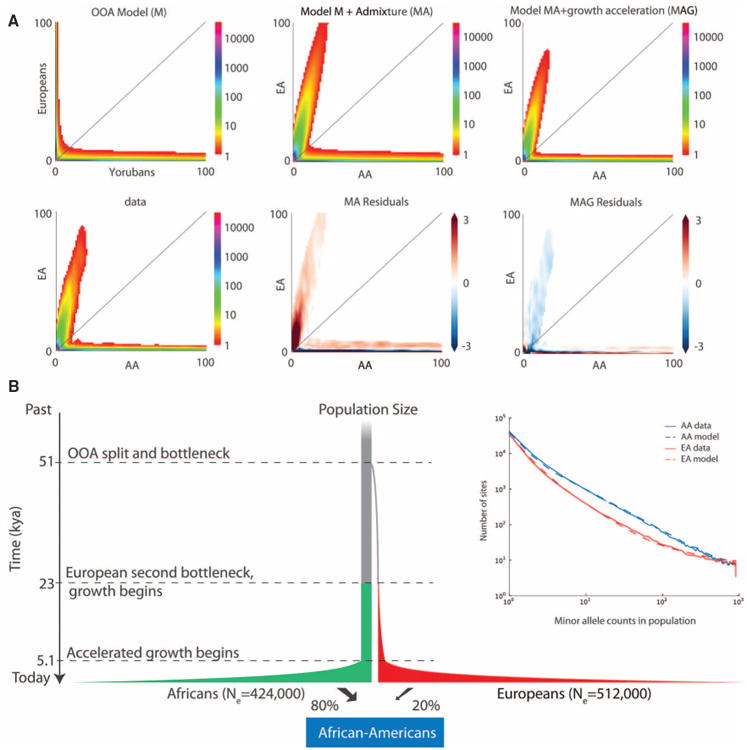
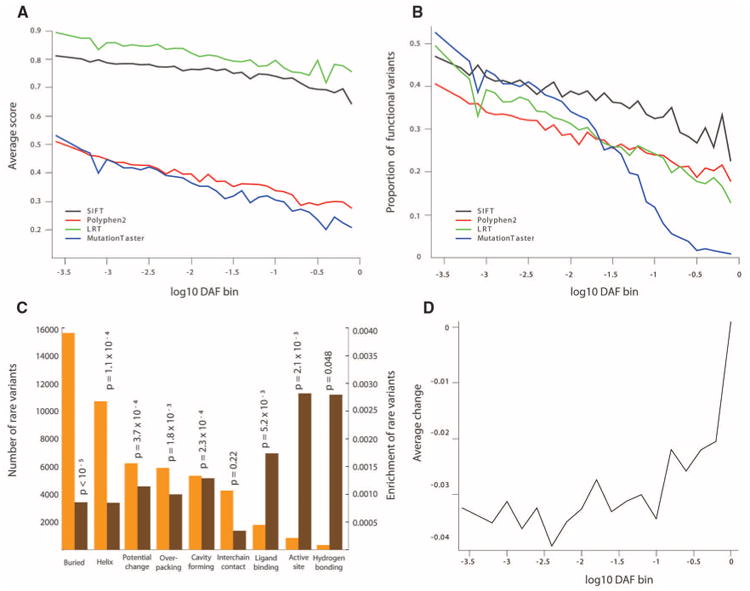
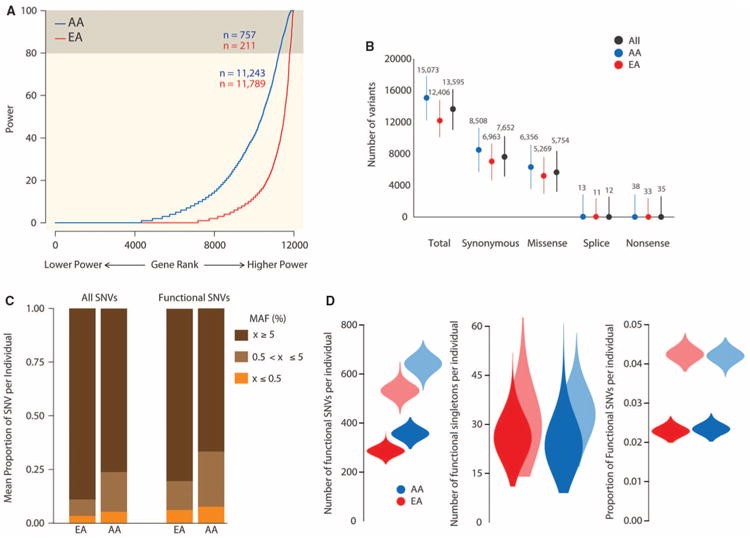
As a first step toward understanding how rare variants contribute to risk for complex diseases, we sequenced 15,585 human protein-coding genes to an average median depth of 111× in 2440 individuals of European (n = 1351) and African (n = 1088) ancestry. We identified over 500,000 single-nucleotide variants (SNVs), the majority of which were rare (86% with a minor allele frequency less than 0.5%), previously unknown (82%), and population-specific (82%). On average, 2.3% of the 13,595 SNVs each person carried were predicted to affect protein function of ~313 genes per genome, and ~95.7% of SNVs predicted to be functionally important were rare. This excess of rare functional variants is due to the combined effects of explosive, recent accelerated population growth and weak purifying selection. Furthermore, we show that large sample sizes will be required to associate rare variants with complex traits.
Figures
Comment in
-
Genetics. Human genetic variation, shared and private.Science. 2012 Jul 6;337(6090):39-40. doi: 10.1126/science.1224528. Science. 2012. PMID: 22767915 No abstract available.
References
-
- Bamshad MJ, et al. Nat Rev Genet. 2011;12:745. - PubMed
-
- International HapMap Consortium. Nature. 2005;437:1299.
Publication types
MeSH terms
Grants and funding
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- RC2 HL-102926/HL/NHLBI NIH HHS/United States
- U01 HG006513/HG/NHGRI NIH HHS/United States
- RC2 HL-102923/HL/NHLBI NIH HHS/United States
- R01 HG003229/HG/NHGRI NIH HHS/United States
- RC2 HL-102925/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- RC2 HL-102924/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL-103010/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
