

Mitochondrial Ca(2+) and neurodegeneration
- PMID: 22608276
- PMCID: PMC3396847
- DOI: 10.1016/j.ceca.2012.04.015
Mitochondrial Ca(2+) and neurodegeneration
Abstract
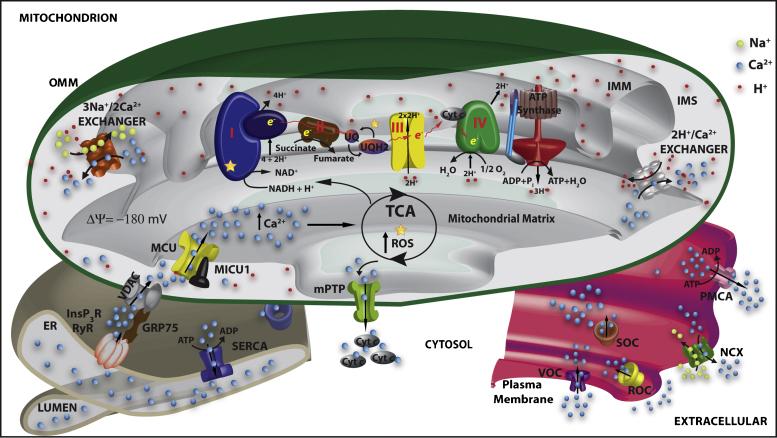
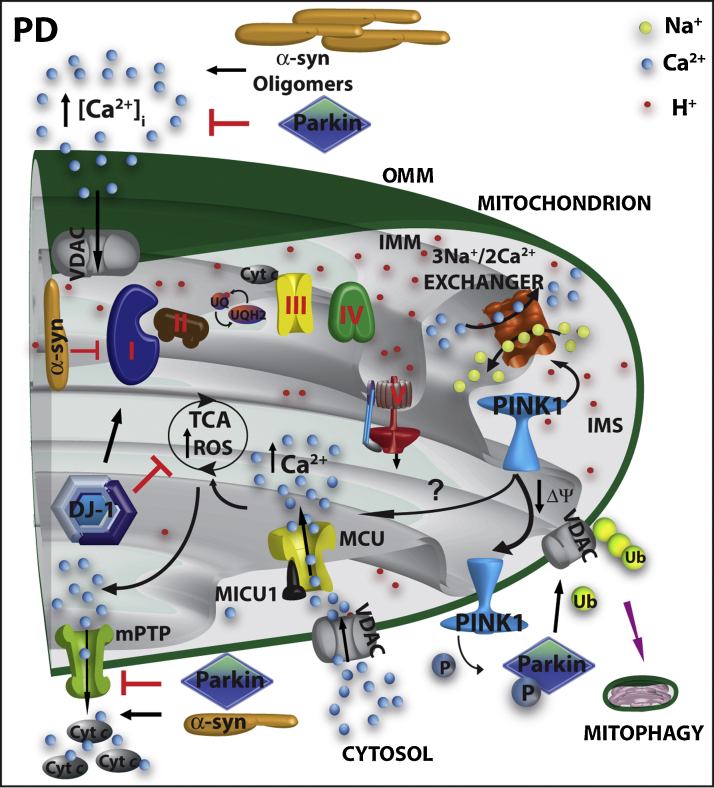
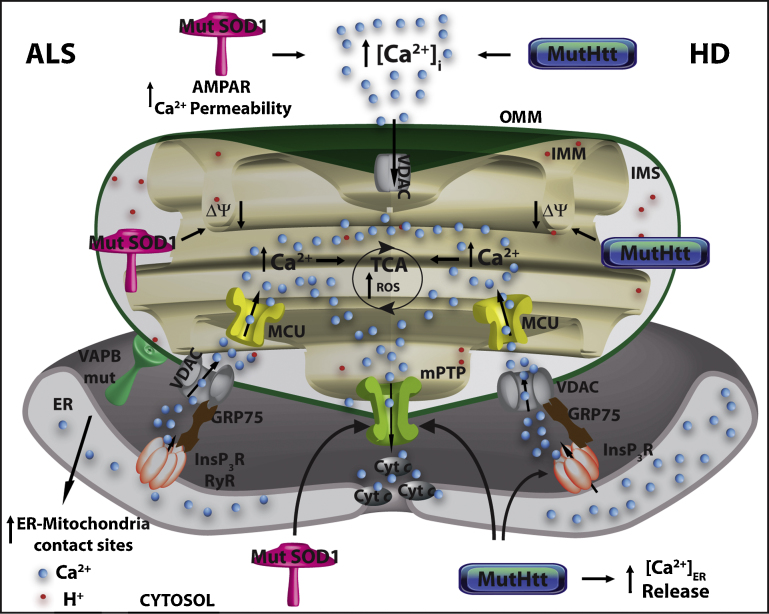
Mitochondria are essential for ensuring numerous fundamental physiological processes such as cellular energy, redox balance, modulation of Ca(2+) signaling and important biosynthetic pathways. They also govern the cell fate by participating in the apoptosis pathway. The mitochondrial shape, volume, number and distribution within the cells are strictly controlled. The regulation of these parameters has an impact on mitochondrial function, especially in the central nervous system, where trafficking of mitochondria is critical to their strategic intracellular distribution, presumably according to local energy demands. Thus, the maintenance of a healthy mitochondrial population is essential to avoid the impairment of the processes they regulate: for this purpose, cells have developed mechanisms involving a complex system of quality control to remove damaged mitochondria, or to renew them. Defects of these processes impair mitochondrial function and lead to disordered cell function, i.e., to a disease condition. Given the standard role of mitochondria in all cells, it might be expected that their dysfunction would give rise to similar defects in all tissues. However, damaged mitochondrial function has pleiotropic effects in multicellular organisms, resulting in diverse pathological conditions, ranging from cardiac and brain ischemia, to skeletal muscle myopathies to neurodegenerative diseases. In this review, we will focus on the relationship between mitochondrial (and cellular) derangements and Ca(2+) dysregulation in neurodegenerative diseases, emphasizing the evidence obtained in genetic models. Common patterns, that recognize the derangement of Ca(2+) and energy control as a causative factor, have been identified: advances in the understanding of the molecular regulation of Ca(2+) homeostasis, and on the ways in which it could become perturbed in neurological disorders, may lead to the development of therapeutic strategies that modulate neuronal Ca(2+) signaling.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures

Similar articles
-
Molecular nature and physiological role of the mitochondrial calcium uniporter channel.Am J Physiol Cell Physiol. 2021 Apr 1;320(4):C465-C482. doi: 10.1152/ajpcell.00502.2020. Epub 2020 Dec 9. Am J Physiol Cell Physiol. 2021. PMID: 33296287 Free PMC article. Review.
-
Overexpression of Mitochondrial Calcium Uniporter Causes Neuronal Death.Oxid Med Cell Longev. 2019 Oct 16;2019:1681254. doi: 10.1155/2019/1681254. eCollection 2019. Oxid Med Cell Longev. 2019. PMID: 31737163 Free PMC article.
-
Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection.Antioxid Redox Signal. 2011 Apr 1;14(7):1275-88. doi: 10.1089/ars.2010.3359. Epub 2010 Oct 6. Antioxid Redox Signal. 2011. PMID: 20615073 Free PMC article. Review.
-
Neuronal calcium homeostasis and dysregulation.Antioxid Redox Signal. 2011 Apr 1;14(7):1261-73. doi: 10.1089/ars.2010.3386. Epub 2010 Nov 30. Antioxid Redox Signal. 2011. PMID: 20626318 Free PMC article. Review.
-
In vivo brain imaging of mitochondrial Ca2+ in neurodegenerative diseases with multiphoton microscopy.Biochim Biophys Acta Mol Cell Res. 2021 May;1868(6):118998. doi: 10.1016/j.bbamcr.2021.118998. Epub 2021 Mar 5. Biochim Biophys Acta Mol Cell Res. 2021. PMID: 33684410 Free PMC article. Review.
Cited by
-
Resveratrol: A potential therapeutic natural polyphenol for neurodegenerative diseases associated with mitochondrial dysfunction.Front Pharmacol. 2022 Sep 16;13:922232. doi: 10.3389/fphar.2022.922232. eCollection 2022. Front Pharmacol. 2022. PMID: 36188541 Free PMC article. Review.
-
Deletion in the N-terminal half of olfactomedin 1 modifies its interaction with synaptic proteins and causes brain dystrophy and abnormal behavior in mice.Exp Neurol. 2013 Dec;250:205-18. doi: 10.1016/j.expneurol.2013.09.019. Epub 2013 Oct 2. Exp Neurol. 2013. PMID: 24095980 Free PMC article.
-
Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity.Nat Med. 2014 Dec;20(12):1427-35. doi: 10.1038/nm.3735. Epub 2014 Nov 24. Nat Med. 2014. PMID: 25419710 Free PMC article.
-
Research progress of mitochondrial dysfunction induced pyroptosis in acute lung injury.Respir Res. 2024 Nov 7;25(1):398. doi: 10.1186/s12931-024-03028-1. Respir Res. 2024. PMID: 39511593 Free PMC article. Review.
-
Dysregulated Ca2+ Homeostasis as a Central Theme in Neurodegeneration: Lessons from Alzheimer's Disease and Wolfram Syndrome.Cells. 2022 Jun 18;11(12):1963. doi: 10.3390/cells11121963. Cells. 2022. PMID: 35741091 Free PMC article. Review.
References
-
- Berridge M.J., Bootman M.D., Roderick H.L. Calcium signalling: dynamics, homeostasis and remodelling. Nature Reviews Molecular Cell Biology. 2003;4:517–529. - PubMed
-
- Berridge M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochimica et Biophysica Acta. 2009;1793:933–940. - PubMed
-
- Brini M., Carafoli E. Calcium pumps in health and disease. Physiological Reviews. 2009;89:1341–1378. - PubMed
-
- Mammucari C., Patron M., Granatiero V., Rizzuto R. Molecules and roles of mitochondrial calcium signaling. Biofactors. 2011;37:219–227. - PubMed
-
- Calì T., Ottolini D., Brini M. Mitochondrial Ca2+ as a key regulator of mitochondrial activities. Advances in mitochondrial medicine. Advances in Experimental Medicine and Biology Series. 2012;942:53–73. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
