

The contribution of CLIP2 haploinsufficiency to the clinical manifestations of the Williams-Beuren syndrome
- PMID: 22608712
- PMCID: PMC3370266
- DOI: 10.1016/j.ajhg.2012.04.020
The contribution of CLIP2 haploinsufficiency to the clinical manifestations of the Williams-Beuren syndrome
Abstract
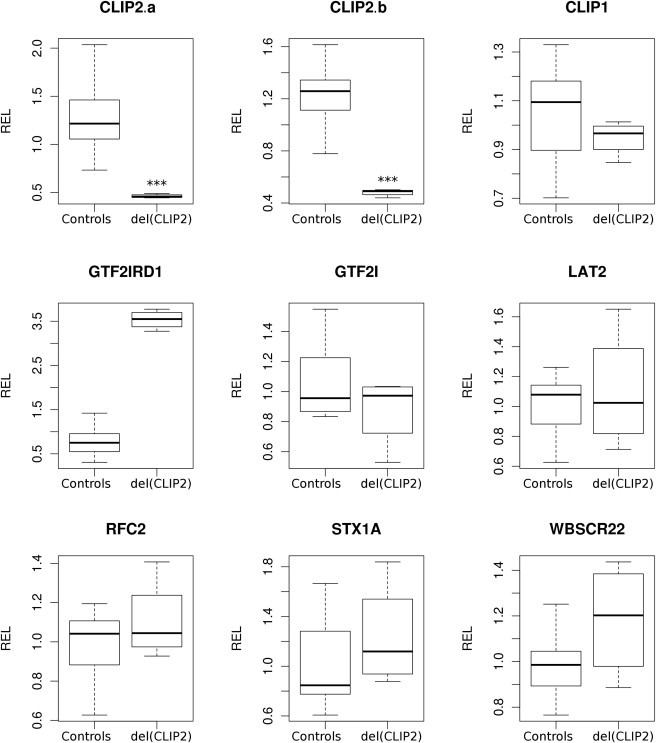
Williams-Beuren syndrome is a rare contiguous gene syndrome, characterized by intellectual disability, facial dysmorphisms, connective-tissue abnormalities, cardiac defects, structural brain abnormalities, and transient infantile hypercalcemia. Genes lying telomeric to RFC2, including CLIP2, GTF2I and GTF2IRD1, are currently thought to be the most likely major contributors to the typical Williams syndrome cognitive profile, characterized by a better-than-expected auditory rote-memory ability, a relative sparing of language capabilities, and a severe visual-spatial constructive impairment. Atypical deletions in the region have helped to establish genotype-phenotype correlations. So far, however, hardly any deletions affecting only a single gene in the disease region have been described. We present here two healthy siblings with a pure, hemizygous deletion of CLIP2. A putative role in the cognitive and behavioral abnormalities seen in Williams-Beuren patients has been suggested for this gene on the basis of observations in a knock-out mouse model. The presented siblings did not show any of the clinical features associated with the syndrome. Cognitive testing showed an average IQ for both and no indication of the Williams syndrome cognitive profile. This shows that CLIP2 haploinsufficiency by itself does not lead to the physical or cognitive characteristics of the Williams-Beuren syndrome, nor does it lead to the Williams syndrome cognitive profile. Although contribution of CLIP2 to the phenotype cannot be excluded when it is deleted in combination with other genes, our results support the hypothesis that GTF2IRD1 and GTF2I are the main genes causing the cognitive defects associated with Williams-Beuren syndrome.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures




Similar articles
-
Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile.J Med Genet. 2010 May;47(5):312-20. doi: 10.1136/jmg.2009.071712. Epub 2009 Nov 5. J Med Genet. 2010. PMID: 19897463
-
Molecular basis of Williams-Beuren syndrome: TFII-I regulated targets involved in craniofacial development.Cleft Palate Craniofac J. 2011 Jan;48(1):109-16. doi: 10.1597/09-093. Epub 2010 Apr 7. Cleft Palate Craniofac J. 2011. PMID: 20500075
-
An atypical 7q11.23 deletion in a normal IQ Williams-Beuren syndrome patient.Eur J Hum Genet. 2010 Jan;18(1):33-8. doi: 10.1038/ejhg.2009.108. Eur J Hum Genet. 2010. PMID: 19568270 Free PMC article.
-
Neurodevelopmental and behavioral issues in Williams syndrome.Curr Psychiatry Rep. 2007 Apr;9(2):165-71. doi: 10.1007/s11920-007-0087-6. Curr Psychiatry Rep. 2007. PMID: 17389129 Review.
-
The contribution of GTF2I haploinsufficiency to Williams syndrome.Mol Cell Probes. 2018 Aug;40:45-51. doi: 10.1016/j.mcp.2017.12.005. Epub 2018 Jan 3. Mol Cell Probes. 2018. PMID: 29305905 Free PMC article. Review.
Cited by
-
Association of GTF2IRD1-GTF2I polymorphisms with neuromyelitis optica spectrum disorders in Han Chinese patients.Neural Regen Res. 2019 Feb;14(2):346-353. doi: 10.4103/1673-5374.244800. Neural Regen Res. 2019. PMID: 30531019 Free PMC article.
-
Genetic contributions to visuospatial cognition in Williams syndrome: insights from two contrasting partial deletion patients.J Neurodev Disord. 2014;6(1):18. doi: 10.1186/1866-1955-6-18. Epub 2014 Jul 15. J Neurodev Disord. 2014. PMID: 25057328 Free PMC article.
-
A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP.Nat Genet. 2014 Apr;46(4):380-4. doi: 10.1038/ng.2899. Epub 2014 Feb 16. Nat Genet. 2014. PMID: 24531329 Free PMC article.
-
Dental-craniofacial manifestation and treatment of rare diseases.Int J Oral Sci. 2019 Feb 20;11(1):9. doi: 10.1038/s41368-018-0041-y. Int J Oral Sci. 2019. PMID: 30783081 Free PMC article. Review.
-
RFC2 may contribute to the pathogenicity of Williams syndrome revealed in a zebrafish model.J Genet Genomics. 2024 Dec;51(12):1389-1403. doi: 10.1016/j.jgg.2024.09.016. Epub 2024 Oct 4. J Genet Genomics. 2024. PMID: 39368701 Free PMC article.
References
-
- Tassabehji M. Williams-Beuren syndrome: A challenge for genotype-phenotype correlations. Hum. Mol. Genet. 2003;12(Spec No 2):R229–R237. - PubMed
-
- Jackowski A.P., Rando K., Maria de Araújo C., Del Cole C.G., Silva I., Tavares de Lacerda A.L. Brain abnormalities in Williams syndrome: a review of structural and functional magnetic resonance imaging findings. Eur. J. Paediatr. Neurol. 2009;13:305–316. - PubMed
-
- Pober B.R. Williams-Beuren syndrome. N. Engl. J. Med. 2010;362:239–252. - PubMed
-
- Mervis C.B., Robinson B.F., Bertrand J., Morris C.A., Klein-Tasman B.P., Armstrong S.C. The Williams syndrome cognitive profile. Brain Cogn. 2000;44:604–628. - PubMed
-
- Rhodes S.M., Riby D.M., Fraser E., Campbell L.E. The extent of working memory deficits associated with Williams syndrome: exploration of verbal and spatial domains and executively controlled processes. Brain Cogn. 2011;77:208–214. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
