

Gene therapy for muscular dystrophy: lessons learned and path forward
- PMID: 22609847
- PMCID: PMC3492936
- DOI: 10.1016/j.neulet.2012.04.078
Gene therapy for muscular dystrophy: lessons learned and path forward
Abstract
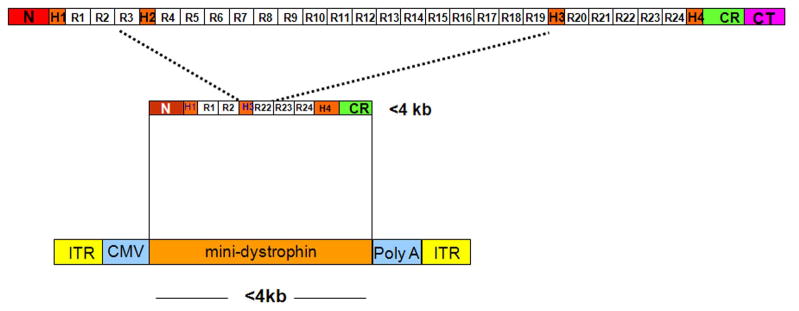
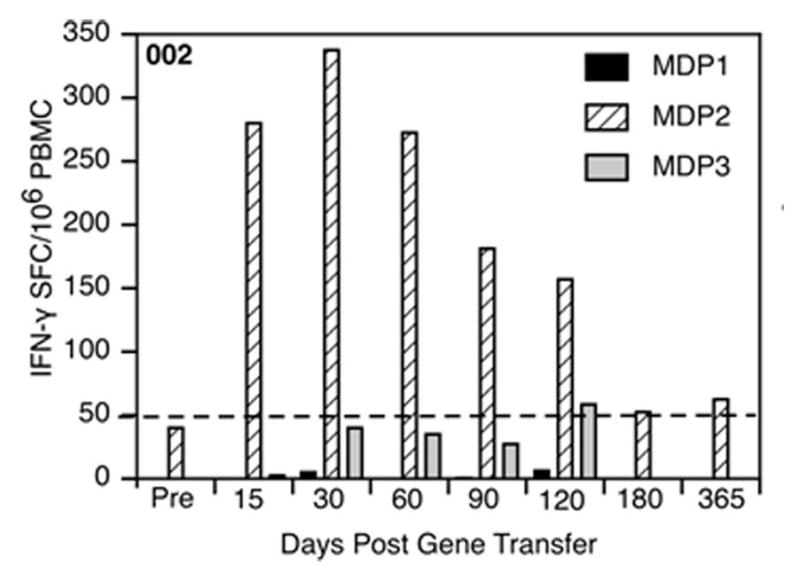
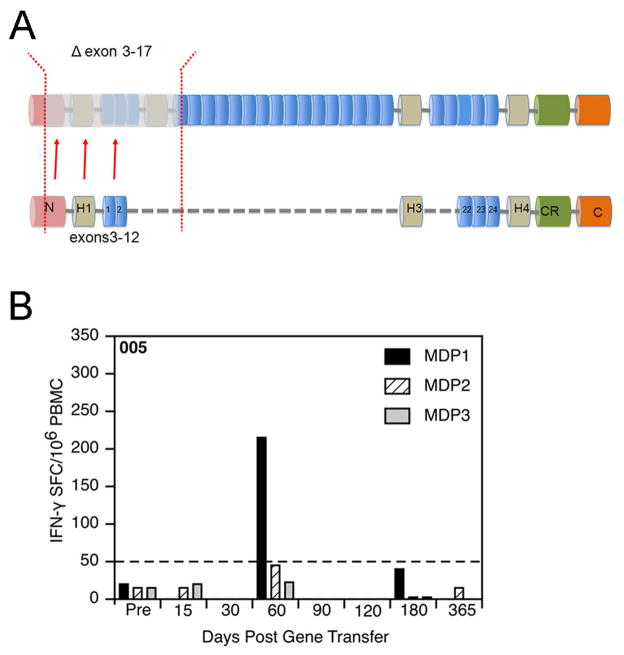
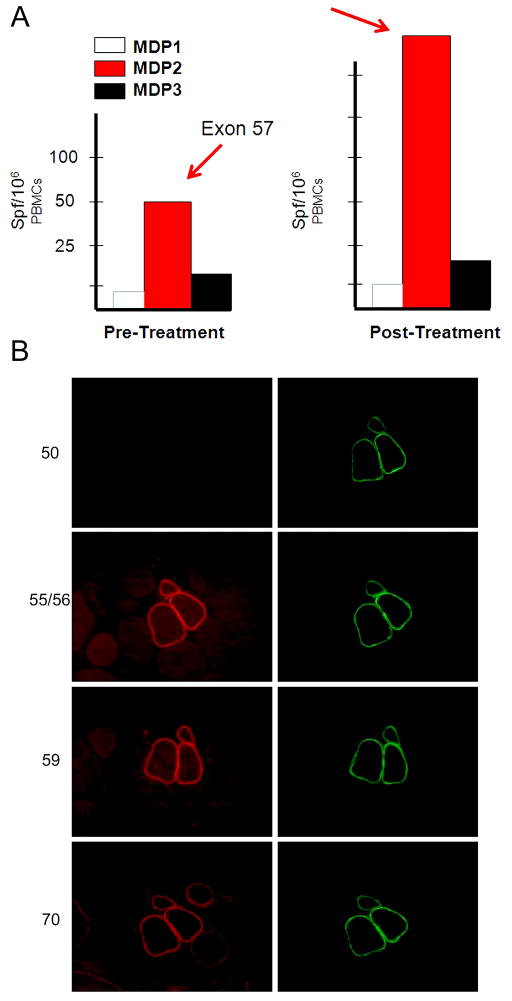
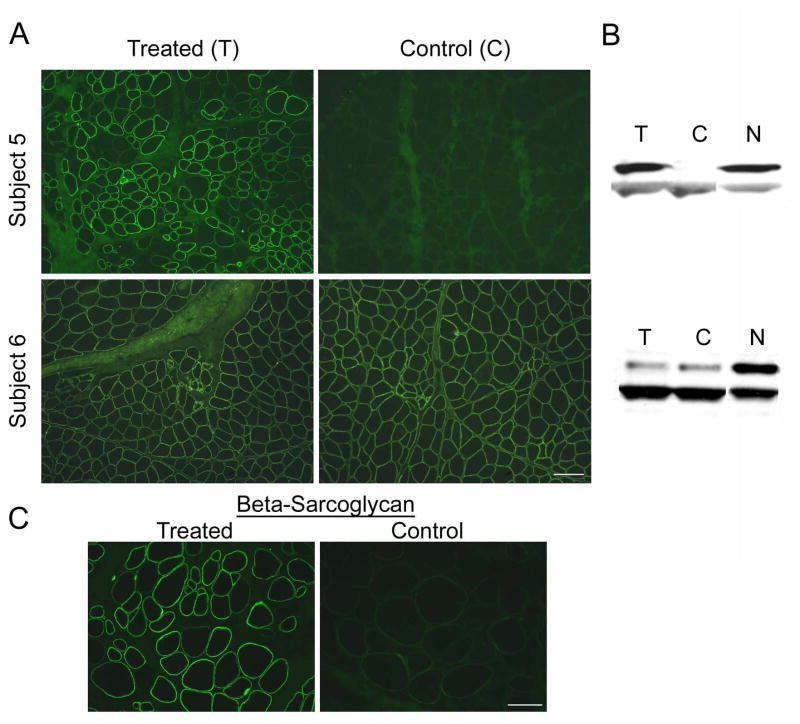
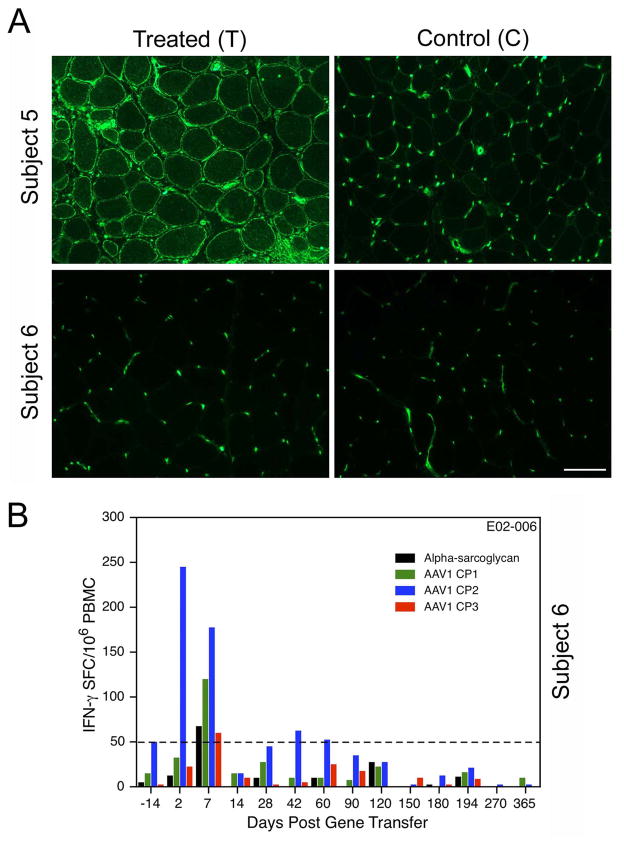
Our Translational Gene Therapy Center has used small molecules for exon skipping and mutation suppression and gene transfer to replace or provide surrogate genes as tools for molecular-based approaches for the treatment of muscular dystrophies. Exon skipping is targeted at the pre-mRNA level allowing one or more exons to be omitted to restore the reading frame. In Duchenne Muscular Dystrophy (DMD), clinical trials have been performed with two different oligomers, a 2'O-methyl-ribo-oligonucleoside-phosphorothioate (2'OMe) and a phosphorodiamidate morpholino (PMO). Both have demonstrated early evidence of efficacy. A second molecular approach involves suppression of stop codons to promote readthrough of the DMD gene. We have been able to establish proof of principle for mutation suppression using the aminoglycoside, gentamicin. A safer, orally administered, alternative agent referred to as Ataluren (PTC124) has been used in clinical trials and is currently under consideration for approval by the FDA. Using a gene therapy approach, we have completed two trials and have initiated a third. For DMD, we used a mini-dystrophin transferred in adeno-associated virus (AAV). In this trial an immune response was seen directed against transgene product, a quite unexpected outcome that will help guide further studies. For limb girdle muscular dystrophy 2D (alpha-sarcoglycan deficiency), the transgene was again transferred using AAV but in this study, a muscle specific creatine kinase promoter controlled gene expression that persisted for six months. A third gene therapy trial has been initiated with transfer of the follistatin gene in AAV directly to the quadriceps muscle. Two diseases with selective quadriceps muscle weakness are undergoing gene transfer including sporadic inclusion body myositis (sIBM) and Becker muscular dystrophy (BMD). Increasing the size and strength of the muscle is the goal of this study. Most importantly, no adverse events have been encountered in any of these clinical trials.
Copyright © 2012 Elsevier Ireland Ltd. All rights reserved.
Figures
References
-
- Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. 2006;16:249–255. - PubMed
-
- Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley R, Miller JP, Province MA. Clinical investigation in Duchenne dystrophy: 2. Determination of the “power” of therapeutic trials based on the natural history. Muscle Nerve. 1983;6:91–103. - PubMed
-
- Chen JY, Clark MJ. Family function in families of children with Duchenne muscular dystrophy. Fam Community Health. 2007;30:296–304. - PubMed
-
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
