

Inhaled hypertonic saline in infants and children younger than 6 years with cystic fibrosis: the ISIS randomized controlled trial
- PMID: 22610452
- PMCID: PMC3586815
- DOI: 10.1001/jama.2012.5214
Inhaled hypertonic saline in infants and children younger than 6 years with cystic fibrosis: the ISIS randomized controlled trial
Abstract
Context: Inhaled hypertonic saline is recommended as therapy for patients 6 years or older with cystic fibrosis (CF), but its efficacy has never been evaluated in patients younger than 6 years with CF.
Objective: To determine if hypertonic saline reduces the rate of protocol-defined pulmonary exacerbations in patients younger than 6 years with CF.
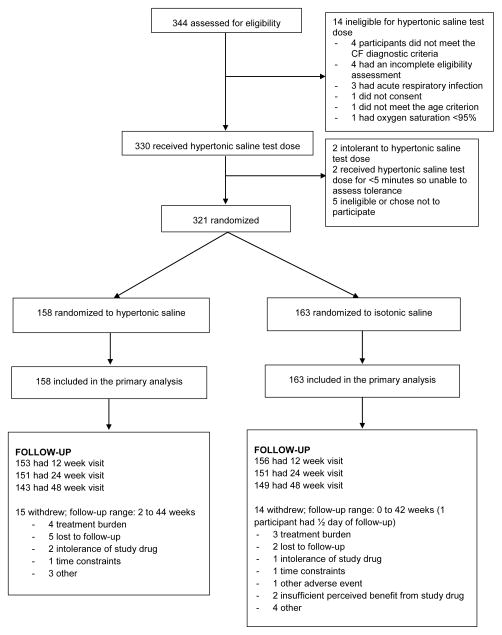
Design, setting, and participants: The Infant Study of Inhaled Saline in Cystic Fibrosis (ISIS), a multicenter, randomized, double-blind, placebo-controlled trial conducted from April 2009 to October 2011 at 30 CF care centers in the United States and Canada. Participants were aged 4 to 60 months and had an established diagnosis of CF. A total of 344 patients were assessed for eligibility; 321 participants were randomized; 29 (9%) withdrew prematurely.
Intervention: The active treatment group (n = 158) received 7% hypertonic saline and the control group (n = 163) received 0.9% isotonic saline, nebulized twice daily for 48 weeks. Both groups received albuterol or levalbuterol prior to each study drug dose.
Main outcome measures: Rate during the 48-week treatment period of protocol-defined pulmonary exacerbations treated with oral, inhaled, or intravenous antibiotics.
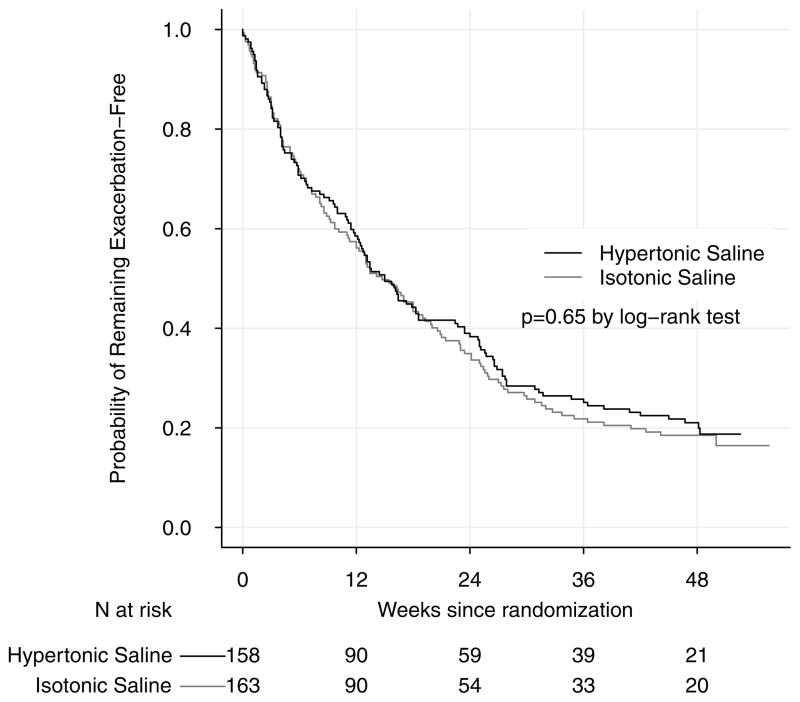
Results: The mean pulmonary exacerbation rate (events per person-year) was 2.3 (95% CI, 2.0-2.5) in the active treatment group and 2.3 (95% CI, 2.1-2.6) in the control group; the adjusted rate ratio was 0.98 (95% CI, 0.84-1.15). Among participants with pulmonary exacerbations, the mean number of total antibiotic treatment days for a pulmonary exacerbation was 60 (95% CI, 49-70) in the active treatment group and 52 (95% CI, 43-61) in the control group. There was no significant difference in secondary end points including height, weight, respiratory rate, oxygen saturation, cough, or respiratory symptom scores. Infant pulmonary function testing performed as an exploratory outcome in a subgroup (n = 73, with acceptable measurements at 2 visits in 45 participants) did not demonstrate significant differences between groups except for the mean change in forced expiratory volume in 0.5 seconds, which was 38 mL (95% CI, 1-76) greater in the active treatment group. Adherence determined by returned study drug ampoules was at least 75% in each group. Adverse event profiles were also similar, with the most common adverse event of moderate or severe severity in each group being cough (39% of active treatment group, 38% of control group).
Conclusion: Among infants and children younger than 6 years with cystic fibrosis, the use of inhaled hypertonic saline compared with isotonic saline did not reduce the rate of pulmonary exacerbations over the course of 48 weeks of treatment.
Trial registration: clinicaltrials.gov Identifier: NCT00709280.
Figures
Comment in
-
Inhaled hypertonic saline in infants and young children with cystic fibrosis.JAMA. 2012 Jun 6;307(21):2316-7. doi: 10.1001/jama.2012.5853. JAMA. 2012. PMID: 22610477 Free PMC article. No abstract available.
References
-
- Ranganathan SC, Stocks J, Dezateux C, et al. The evolution of airway function in early childhood following clinical diagnosis of cystic fibrosis. Am J Respir Crit Care Med. 2004;169:928–33. - PubMed
-
- Davis SD, Fordham LA, Brody AS, et al. Computed tomography reflects lower airway inflammation and tracks changes in early cystic fibrosis. Am J Respir Crit Care Med. 2007;175:943–50. - PubMed
-
- Mott LS, Park J, Murray CP, et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax. 2011 - PubMed
-
- Boucher RC. Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med. 2007;261:5–16. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
