

Proteome sampling by the HLA class I antigen processing pathway
- PMID: 22615552
- PMCID: PMC3355062
- DOI: 10.1371/journal.pcbi.1002517
Proteome sampling by the HLA class I antigen processing pathway
Abstract
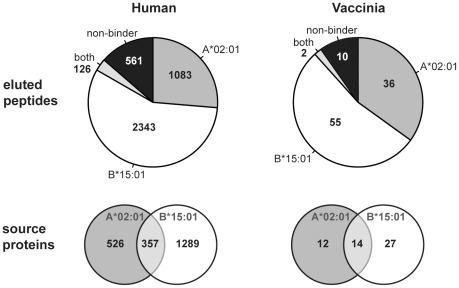
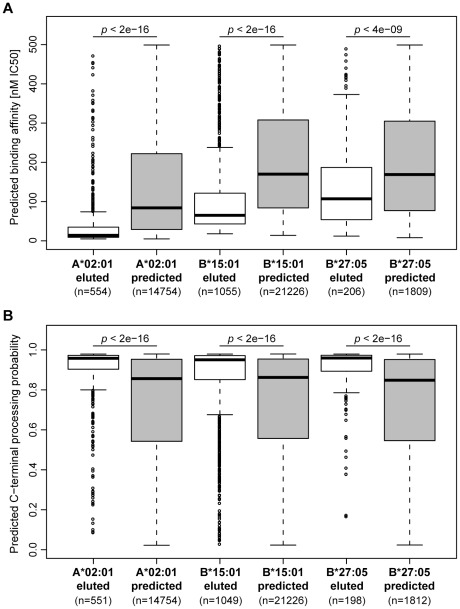
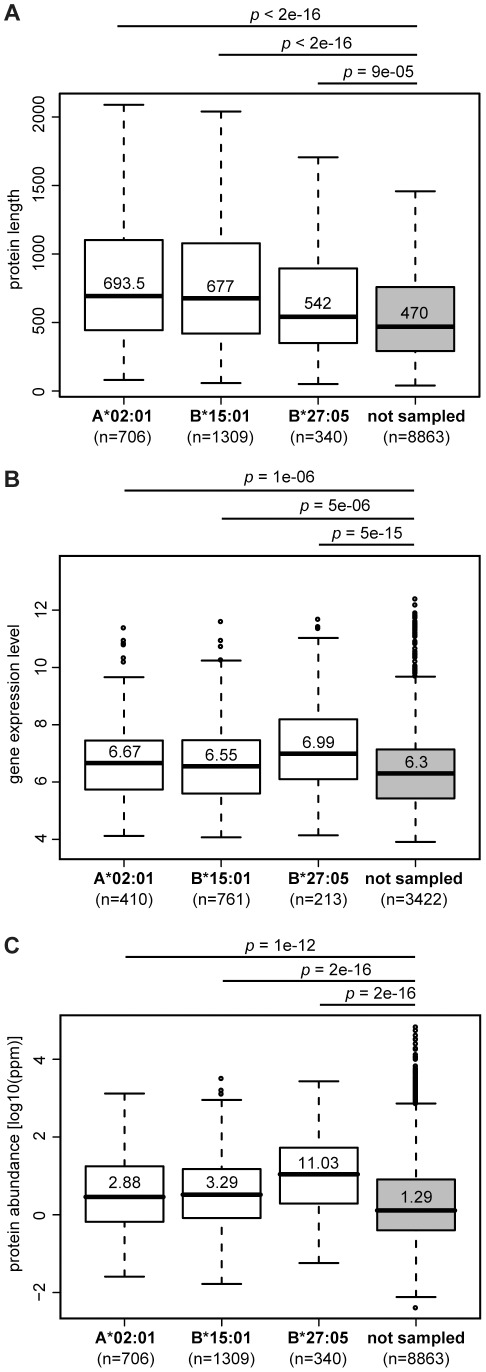
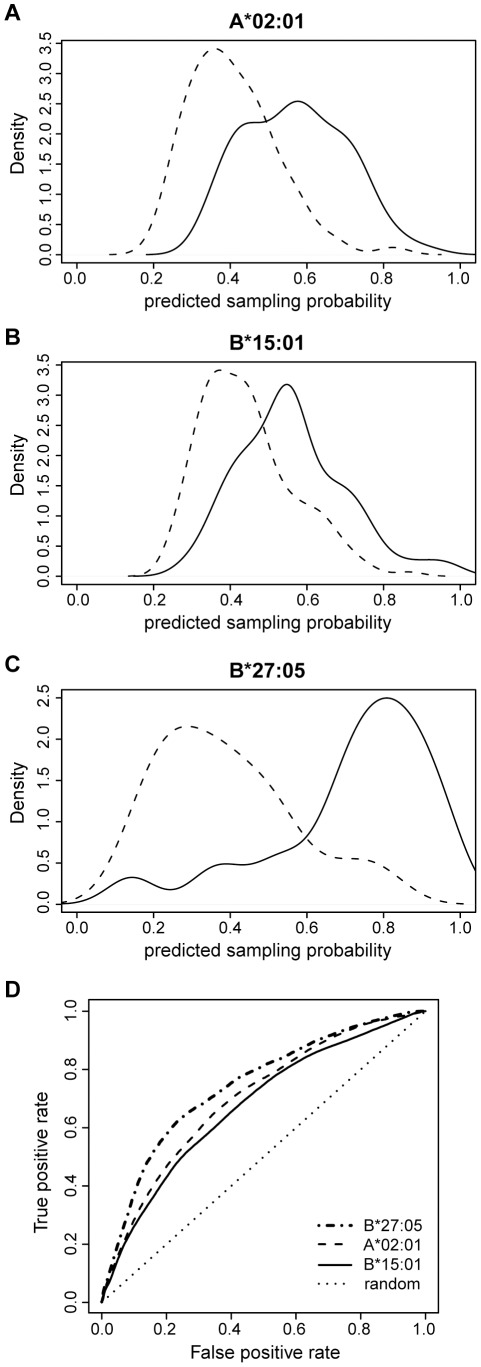
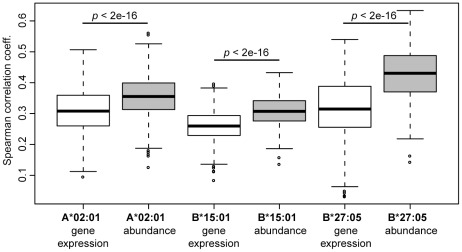
The peptide repertoire that is presented by the set of HLA class I molecules of an individual is formed by the different players of the antigen processing pathway and the stringent binding environment of the HLA class I molecules. Peptide elution studies have shown that only a subset of the human proteome is sampled by the antigen processing machinery and represented on the cell surface. In our study, we quantified the role of each factor relevant in shaping the HLA class I peptide repertoire by combining peptide elution data, in silico predictions of antigen processing and presentation, and data on gene expression and protein abundance. Our results indicate that gene expression level, protein abundance, and rate of potential binding peptides per protein have a clear impact on sampling probability. Furthermore, once a protein is available for the antigen processing machinery in sufficient amounts, C-terminal processing efficiency and binding affinity to the HLA class I molecule determine the identity of the presented peptides. Having studied the impact of each of these factors separately, we subsequently combined all factors in a logistic regression model in order to quantify their relative impact. This model demonstrated the superiority of protein abundance over gene expression level in predicting sampling probability. Being able to discriminate between sampled and non-sampled proteins to a significant degree, our approach can potentially be used to predict the sampling probability of self proteins and of pathogen-derived proteins, which is of importance for the identification of autoimmune antigens and vaccination targets.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. - PubMed
-
- Huseby ES, White J, Crawford F, Vass T, Becker D, et al. How the T cell repertoire becomes peptide and MHC specific. Cell. 2005;122:247–260. - PubMed
-
- Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol. 2004;22:765–787. - PubMed
-
- Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol. 2002;20:217–251. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
