

Replacing and additive horizontal gene transfer in Streptococcus
- PMID: 22617954
- PMCID: PMC3472495
- DOI: 10.1093/molbev/mss138
Replacing and additive horizontal gene transfer in Streptococcus
Abstract
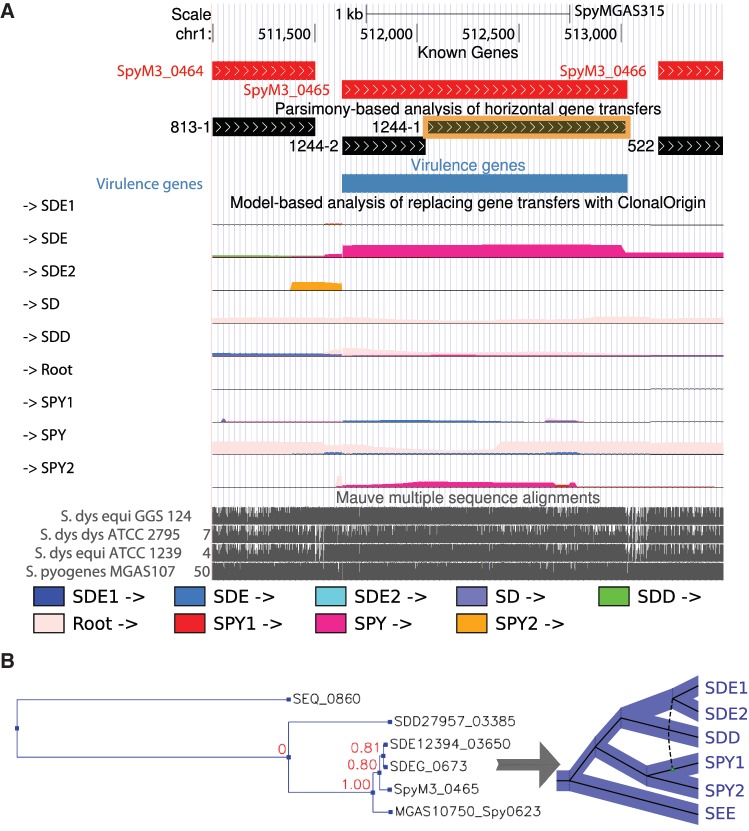
The prominent role of Horizontal Gene Transfer (HGT) in the evolution of bacteria is now well documented, but few studies have differentiated between evolutionary events that predominantly cause genes in one lineage to be replaced by homologs from another lineage ("replacing HGT") and events that result in the addition of substantial new genomic material ("additive HGT"). Here in, we make use of the distinct phylogenetic signatures of replacing and additive HGTs in a genome-wide study of the important human pathogen Streptococcus pyogenes (SPY) and its close relatives S. dysgalactiae subspecies equisimilis (SDE) and S. dysgalactiae subspecies dysgalactiae (SDD). Using recently developed statistical models and computational methods, we find evidence for abundant gene flow of both kinds within each of the SPY and SDE clades and of reduced levels of exchange between SPY and SDD. In addition, our analysis strongly supports a pronounced asymmetry in SPY-SDE gene flow, favoring the SPY-to-SDE direction. This finding is of particular interest in light of the recent increase in virulence of pathogenic SDE. We find much stronger evidence for SPY-SDE gene flow among replacing than among additive transfers, suggesting a primary influence from homologous recombination between co-occurring SPY and SDE cells in human hosts. Putative virulence genes are correlated with transfer events, but this correlation is found to be driven by additive, not replacing, HGTs. The genes affected by additive HGTs are enriched for functions having to do with transposition, recombination, and DNA integration, consistent with previous findings, whereas replacing HGTs seen to influence a more diverse set of genes. Additive transfers are also found to be associated with evidence of positive selection. These findings shed new light on the manner in which HGT has shaped pathogenic bacterial genomes.
Figures




References
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
