

TaxMan: a server to trim rRNA reference databases and inspect taxonomic coverage
- PMID: 22618877
- PMCID: PMC3394339
- DOI: 10.1093/nar/gks418
TaxMan: a server to trim rRNA reference databases and inspect taxonomic coverage
Abstract
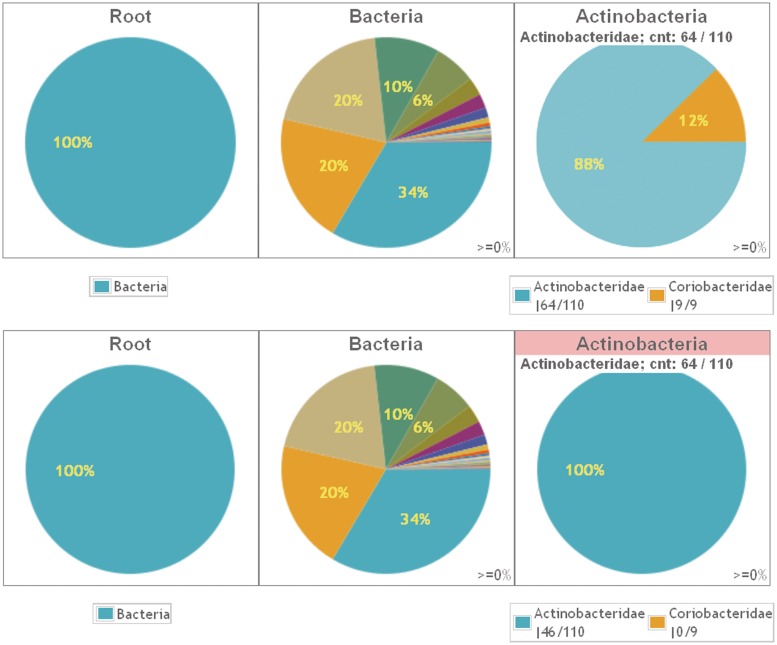
Amplicon sequencing of the hypervariable regions of the small subunit ribosomal RNA gene is a widely accepted method for identifying the members of complex bacterial communities. Several rRNA gene sequence reference databases can be used to assign taxonomic names to the sequencing reads using BLAST, USEARCH, GAST or the RDP classifier. Next-generation sequencing methods produce ample reads, but they are short, currently ∼100-450 nt (depending on the technology), as compared to the full rRNA gene of ∼1550 nt. It is important, therefore, to select the right rRNA gene region for sequencing. The primers should amplify the species of interest and the hypervariable regions should differentiate their taxonomy. Here, we introduce TaxMan: a web-based tool that trims reference sequences based on user-selected primer pairs and returns an assessment of the primer specificity by taxa. It allows interactive plotting of taxa, both amplified and missed in silico by the primers used. Additionally, using the trimmed sequences improves the speed of sequence matching algorithms. The smaller database greatly improves run times (up to 98%) and memory usage, not only of similarity searching (BLAST), but also of chimera checking (UCHIME) and of clustering the reads (UCLUST). TaxMan is available at http://www.ibi.vu.nl/programs/taxmanwww/.
Figures

References
-
- Röling WFM, Head IM. Prokaryotic systematics: PCR and sequence analysis of amplified 16S rRNA genes. In: Osborn AM, Smith CJ, editors. Molecular Microbial Ecology. New York: Taylor & Francis Group; 2005. pp. 25–56.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
