

Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23)
- PMID: 22619378
- PMCID: PMC3406759
- DOI: 10.1093/hmg/dds194
Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23)
Abstract
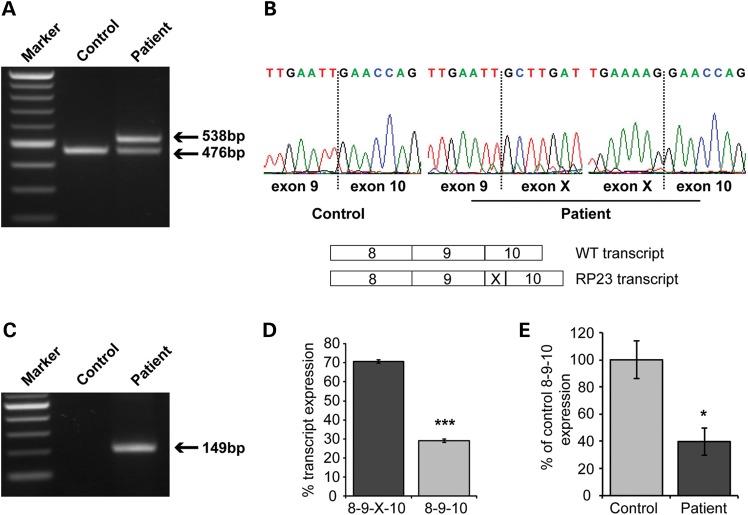
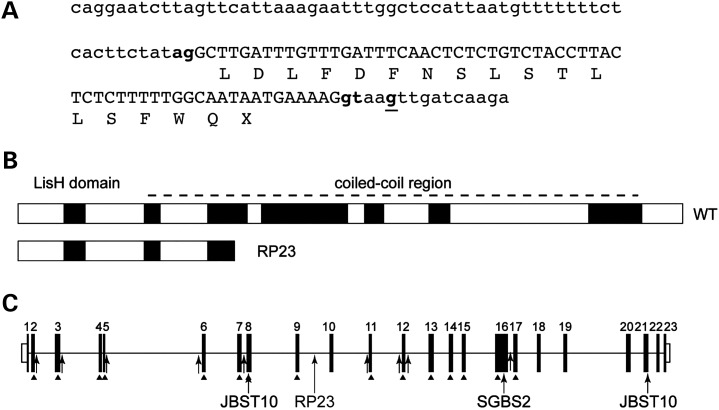
X-linked retinitis pigmentosa (XLRP) is genetically heterogeneous with two causative genes identified, RPGR and RP2. We previously mapped a locus for a severe form of XLRP, RP23, to a 10.71 Mb interval on Xp22.31-22.13 containing 62 genes. Candidate gene screening failed to identify a causative mutation, so we adopted targeted genomic next-generation sequencing of the disease interval to determine the molecular cause of RP23. No coding variants or variants within or near splice sites were identified. In contrast, a variant deep within intron 9 of OFD1 increased the splice site prediction score 4 bp upstream of the variant. Mutations in OFD1 cause the syndromic ciliopathies orofaciodigital syndrome-1, which is male lethal, Simpson-Golabi-Behmel syndrome type 2 and Joubert syndrome. We tested the effect of the IVS9+706A>G variant on OFD1 splicing in vivo. In RP23 patient-derived RNA, we detected an OFD1 transcript with the insertion of a cryptic exon spliced between exons 9 and 10 causing a frameshift, p.N313fs.X330. Correctly spliced OFD1 was also detected in patient-derived RNA, although at reduced levels (39%), hence the mutation is not male lethal. Our data suggest that photoreceptors are uniquely susceptible to reduced expression of OFD1 and that an alternative disease mechanism can cause XLRP. This disease mechanism of reduced expression for a syndromic ciliopathy gene causing isolated retinal degeneration is reminiscent of CEP290 intronic mutations that cause Leber congenital amaurosis, and we speculate that reduced dosage of correctly spliced ciliopathy genes may be a common disease mechanism in retinal degenerations.
Figures

References
-
- Meindl A., Dry K., Herrmann K., Manson F., Ciccodicola A., Edgar A., Carvalho M.R., Achatz H., Hellebrand H., Lennon A., et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3) Nat. Genet. 1996;13:35–42. - PubMed
-
- Schwahn U., Lenzner S., Dong J., Feil S., Hinzmann B., van Duijnhoven G., Kirschner R., Hemberger M., Bergen A.A., Rosenberg T., et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat. Genet. 1998;19:327–332. - PubMed
-
- Shu X., Black G.C., Rice J.M., Hart-Holden N., Jones A., O'Grady A., Ramsden S., Wright A.F. RPGR mutation analysis and disease: an update. Hum. Mutat. 2007;28:322–328. - PubMed
-
- Vervoort R., Lennon A., Bird A.C., Tulloch B., Axton R., Miano M.G., Meindl A., Meitinger T., Ciccodicola A., Wright A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000;25:462–466. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
