

Arenavirus nucleoproteins prevent activation of nuclear factor kappa B
- PMID: 22623788
- PMCID: PMC3421694
- DOI: 10.1128/JVI.07240-11
Arenavirus nucleoproteins prevent activation of nuclear factor kappa B
Abstract
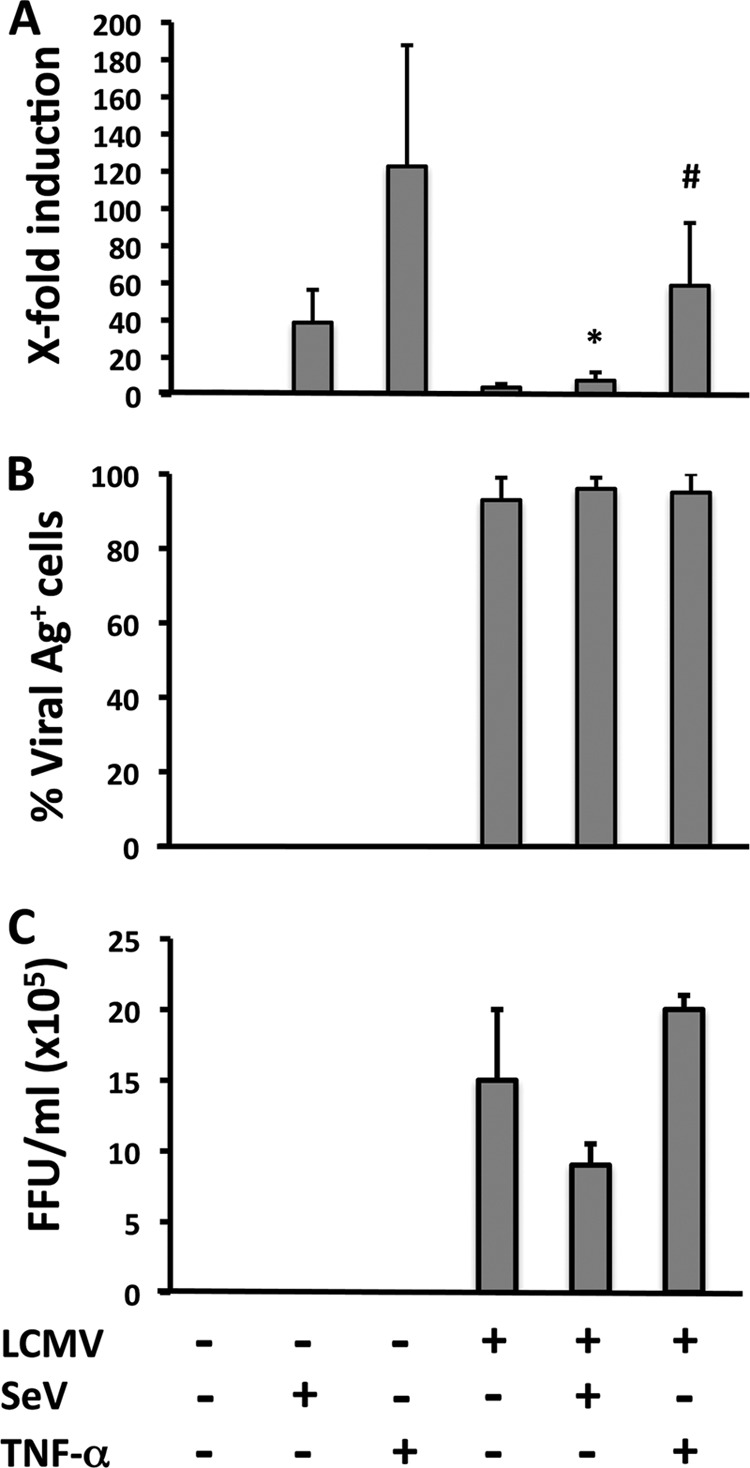
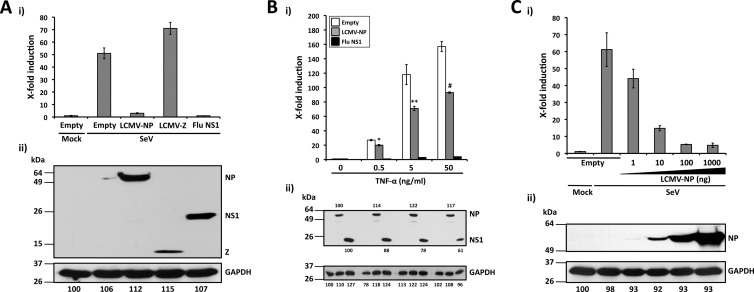
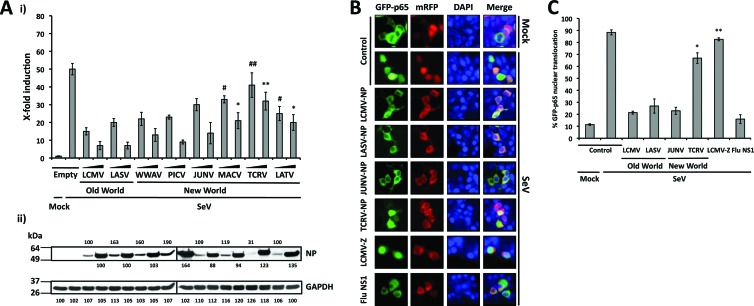
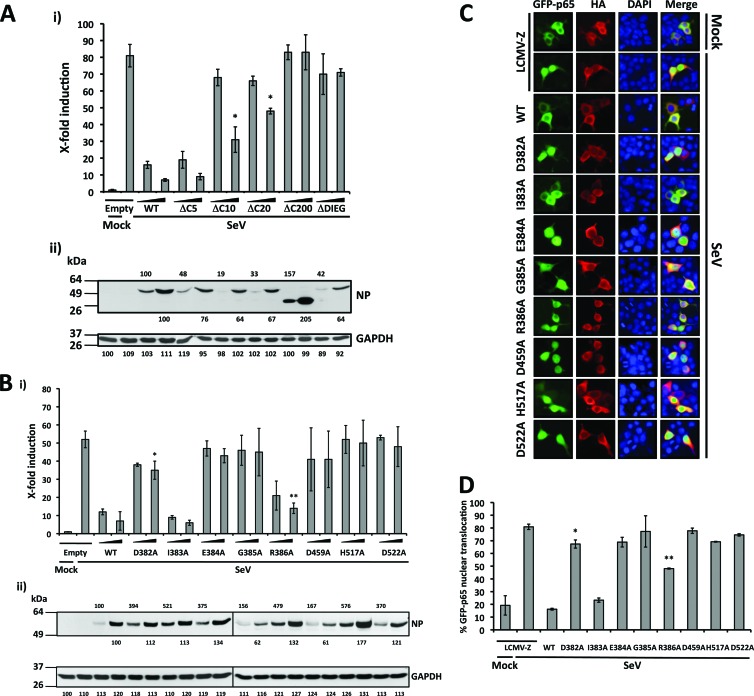
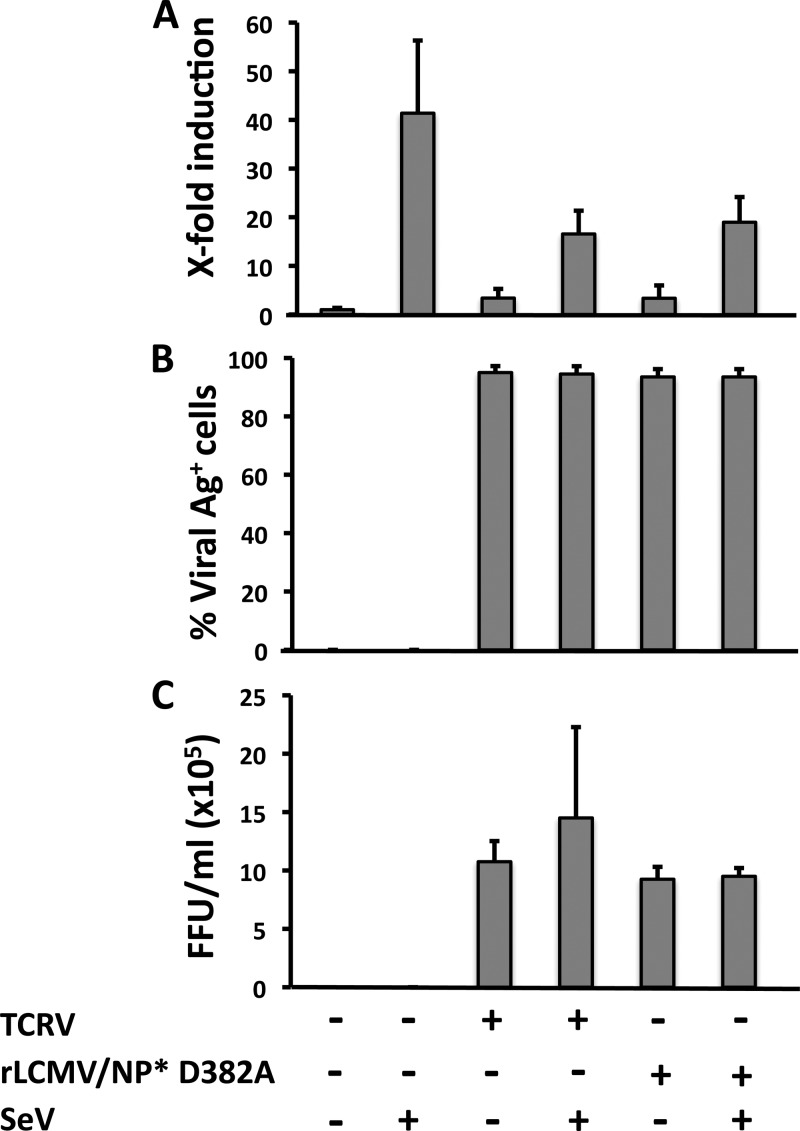
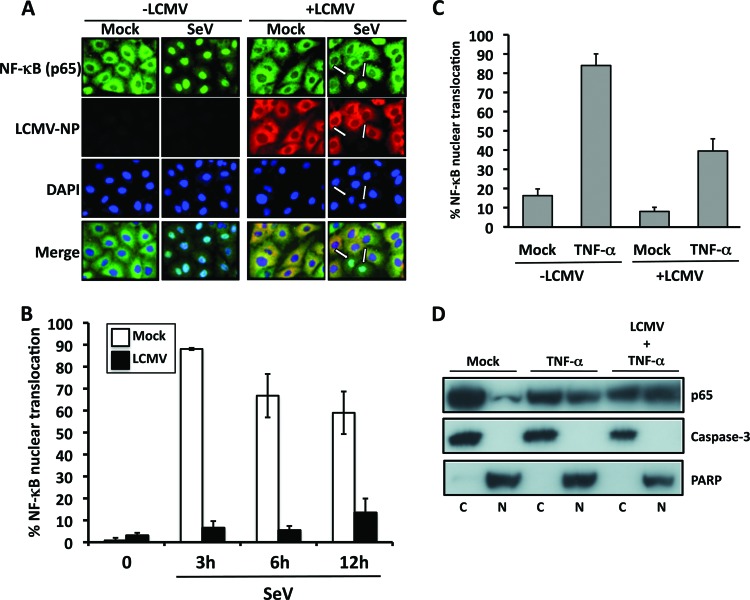
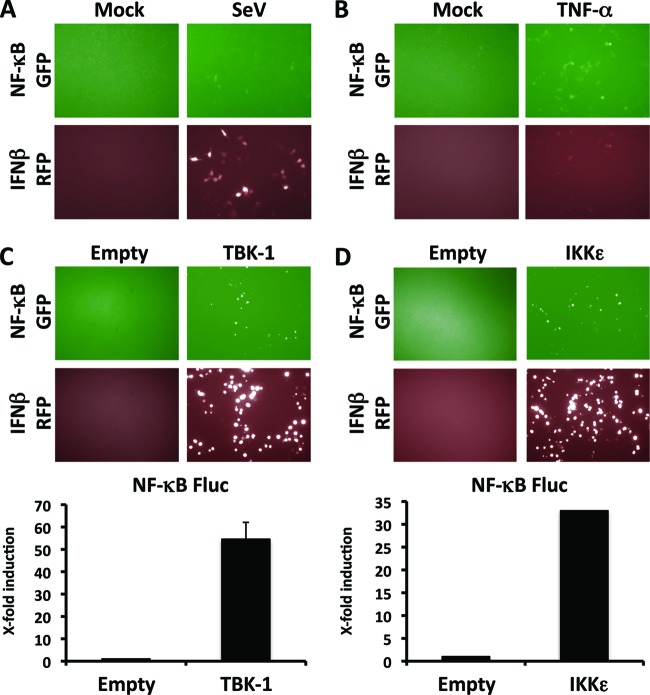
Arenaviruses include several causative agents of hemorrhagic fever (HF) disease in humans that are associated with high morbidity and significant mortality. Morbidity and lethality associated with HF arenaviruses are believed to involve the dysregulation of the host innate immune and inflammatory responses that leads to impaired development of protective and efficient immunity. The molecular mechanisms underlying this dysregulation are not completely understood, but it is suggested that viral infection leads to disruption of early host defenses and contributes to arenavirus pathogenesis in humans. We demonstrate in the accompanying paper that the prototype member in the family, lymphocytic choriomeningitis virus (LCMV), disables the host innate defense by interfering with type I interferon (IFN-I) production through inhibition of the interferon regulatory factor 3 (IRF3) activation pathway and that the viral nucleoprotein (NP) alone is responsible for this inhibitory effect (C. Pythoud, W. W. Rodrigo, G. Pasqual, S. Rothenberger, L. Martínez-Sobrido, J. C. de la Torre, and S. Kunz, J. Virol. 86:7728-7738, 2012). In this report, we show that LCMV-NP, as well as NPs encoded by representative members of both Old World (OW) and New World (NW) arenaviruses, also inhibits the nuclear translocation and transcriptional activity of the nuclear factor kappa B (NF-κB). Similar to the situation previously reported for IRF3, Tacaribe virus NP (TCRV-NP) does not inhibit NF-κB nuclear translocation and transcriptional activity to levels comparable to those seen with other members in the family. Altogether, our findings demonstrate that arenavirus infection inhibits NF-κB-dependent innate immune and inflammatory responses, possibly playing a key role in the pathogenesis and virulence of arenavirus.
Figures
References
-
- Borrow P, Oldstone MB. 1994. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology 198:1–9 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
