

Recurrent hemizygous deletions in cancers may optimize proliferative potential
- PMID: 22628553
- PMCID: PMC4027969
- DOI: 10.1126/science.1219580
Recurrent hemizygous deletions in cancers may optimize proliferative potential
Abstract
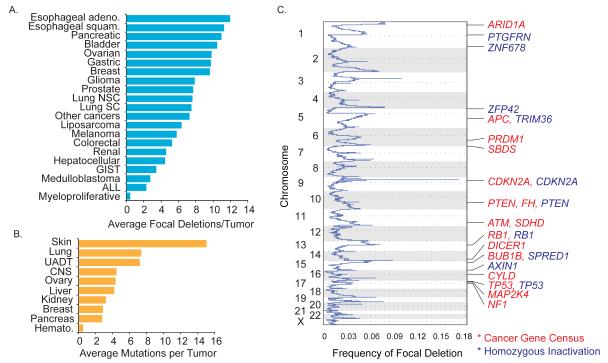
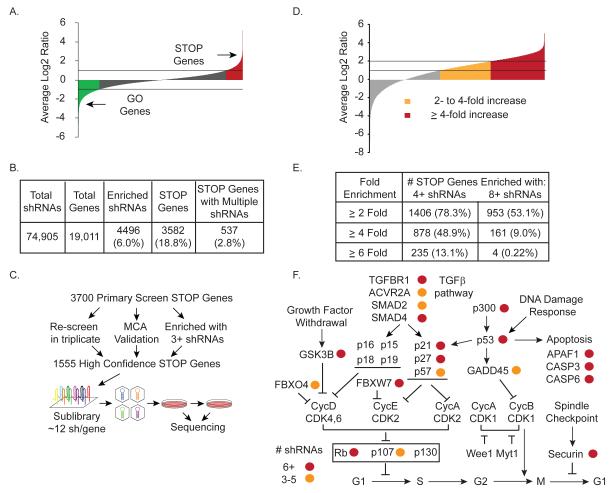
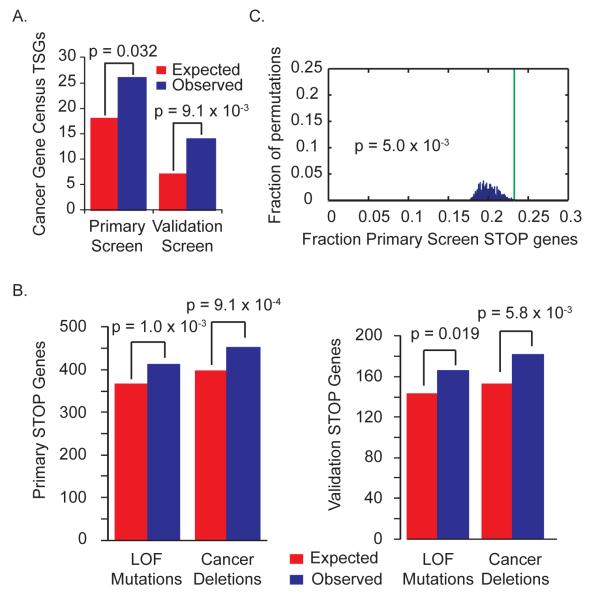
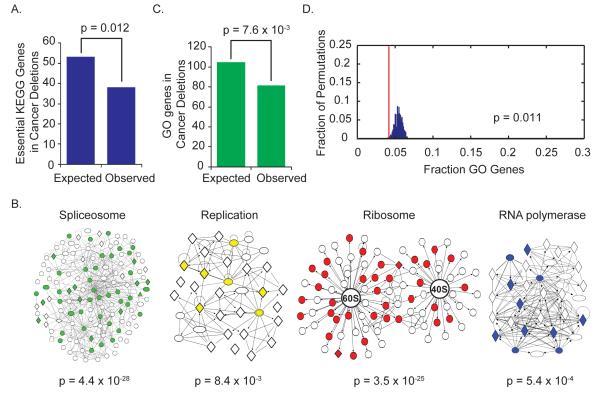
Tumors exhibit numerous recurrent hemizygous focal deletions that contain no known tumor suppressors and are poorly understood. To investigate whether these regions contribute to tumorigenesis, we searched genetically for genes with cancer-relevant properties within these hemizygous deletions. We identified STOP and GO genes, which negatively and positively regulate proliferation, respectively. STOP genes include many known tumor suppressors, whereas GO genes are enriched for essential genes. Analysis of their chromosomal distribution revealed that recurring deletions preferentially overrepresent STOP genes and underrepresent GO genes. We propose a hypothesis called the cancer gene island model, whereby gene islands encompassing high densities of STOP genes and low densities of GO genes are hemizygously deleted to maximize proliferative fitness through cumulative haploinsufficiencies. Because hundreds to thousands of genes are hemizygously deleted per tumor, this mechanism may help to drive tumorigenesis across many cancer types.
Figures
Comment in
-
Tumorigenesis. Better by half.Nat Rev Cancer. 2012 Jun 22;12(7):450-1. doi: 10.1038/nrc3307. Nat Rev Cancer. 2012. PMID: 22722395
-
Cancer. Haploinsufficient gene selection in cancer.Science. 2012 Jul 6;337(6090):47-8. doi: 10.1126/science.1224806. Science. 2012. PMID: 22767920 No abstract available.
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144:646. - PubMed
-
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004 Aug;10:789. - PubMed
-
- Garraway LA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005 Jul 7;436:117. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
