

Ferroptosis: an iron-dependent form of nonapoptotic cell death
- PMID: 22632970
- PMCID: PMC3367386
- DOI: 10.1016/j.cell.2012.03.042
Ferroptosis: an iron-dependent form of nonapoptotic cell death
Abstract
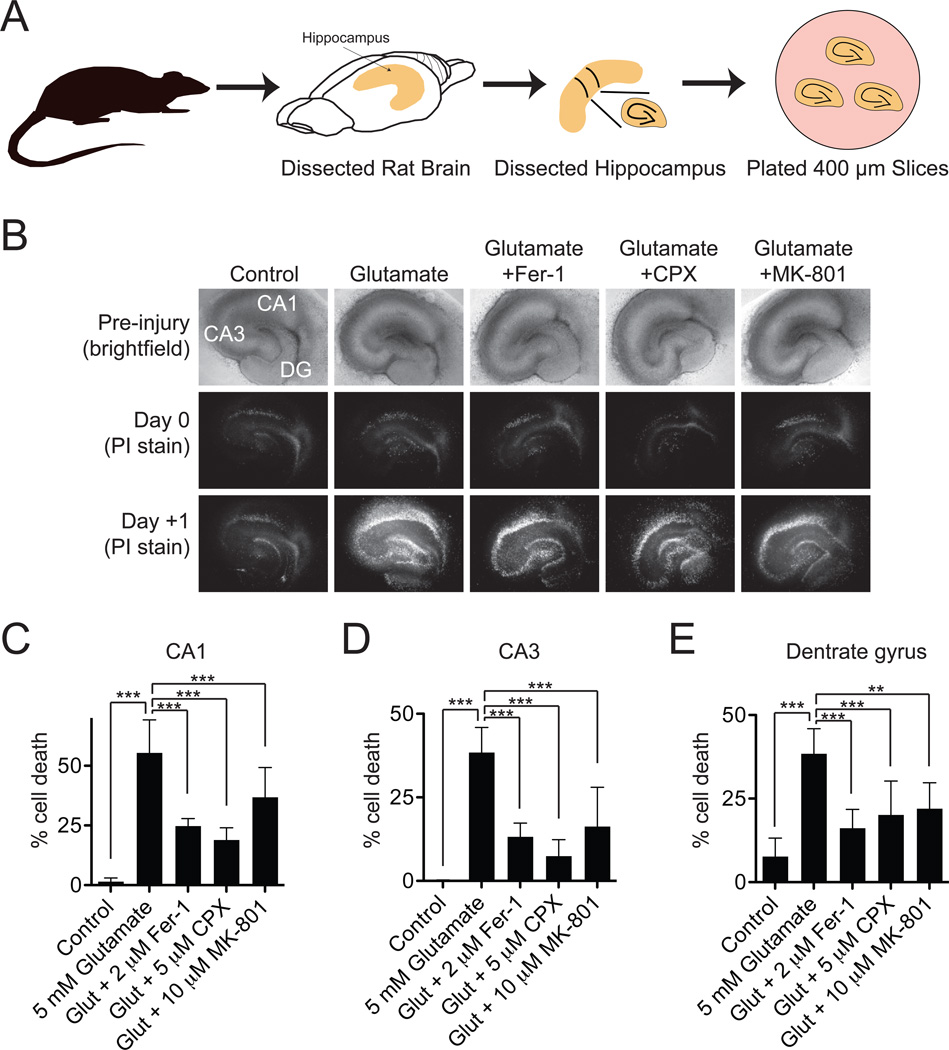
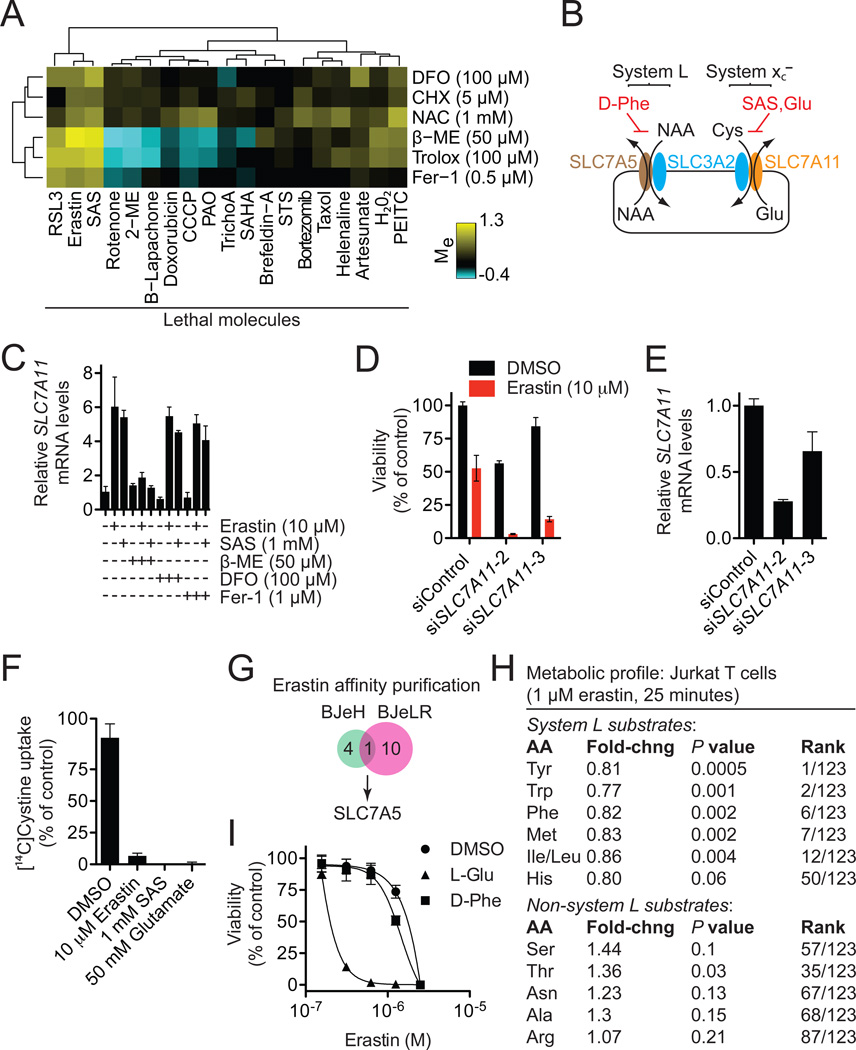
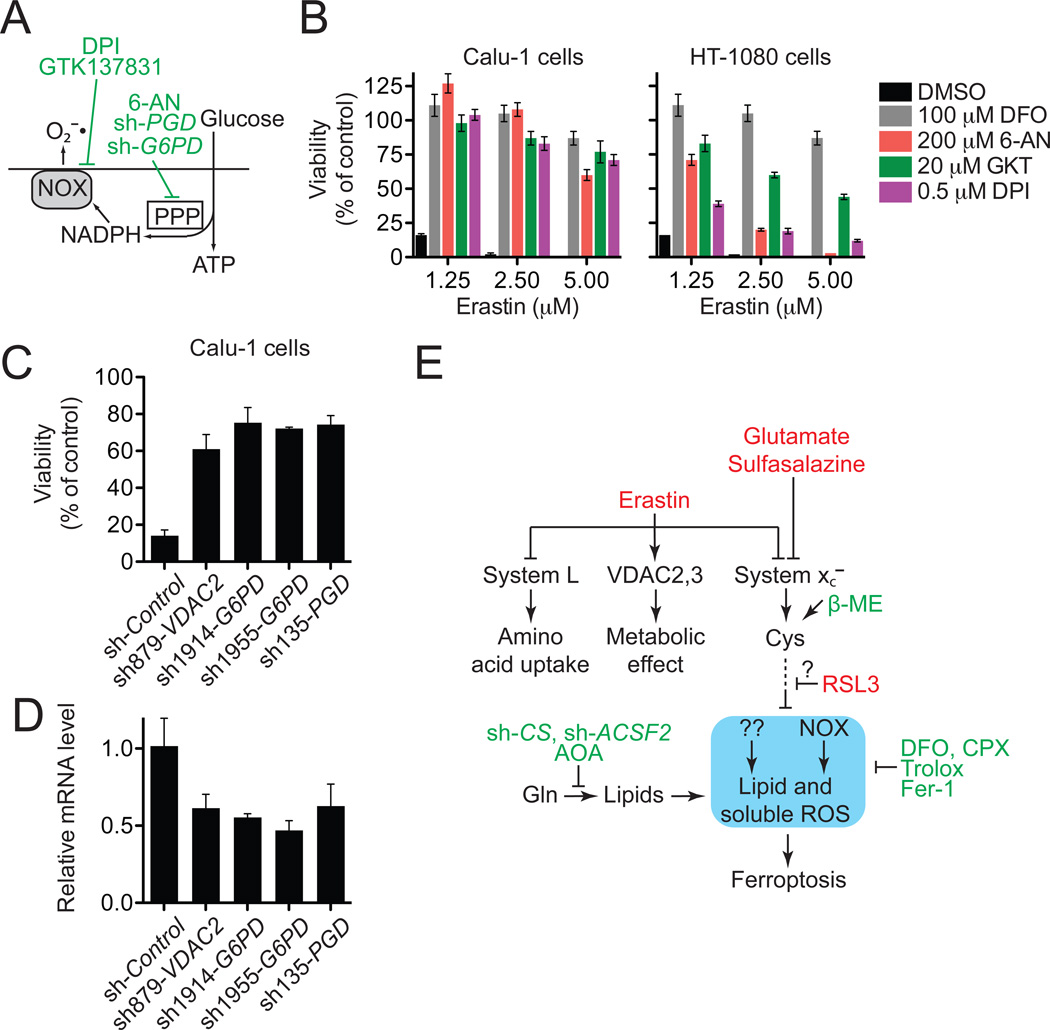
Nonapoptotic forms of cell death may facilitate the selective elimination of some tumor cells or be activated in specific pathological states. The oncogenic RAS-selective lethal small molecule erastin triggers a unique iron-dependent form of nonapoptotic cell death that we term ferroptosis. Ferroptosis is dependent upon intracellular iron, but not other metals, and is morphologically, biochemically, and genetically distinct from apoptosis, necrosis, and autophagy. We identify the small molecule ferrostatin-1 as a potent inhibitor of ferroptosis in cancer cells and glutamate-induced cell death in organotypic rat brain slices, suggesting similarities between these two processes. Indeed, erastin, like glutamate, inhibits cystine uptake by the cystine/glutamate antiporter (system x(c)(-)), creating a void in the antioxidant defenses of the cell and ultimately leading to iron-dependent, oxidative death. Thus, activation of ferroptosis results in the nonapoptotic destruction of certain cancer cells, whereas inhibition of this process may protect organisms from neurodegeneration.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
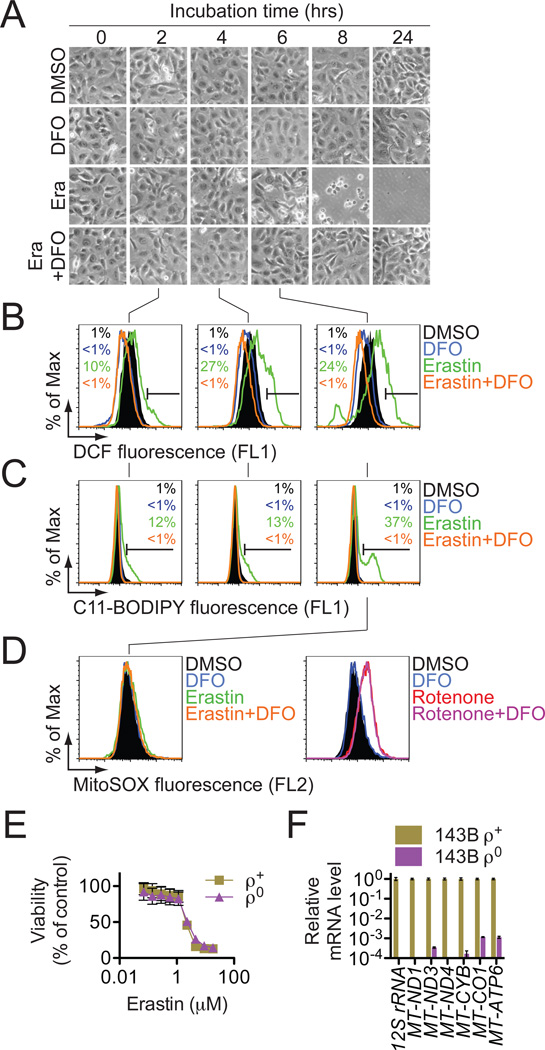
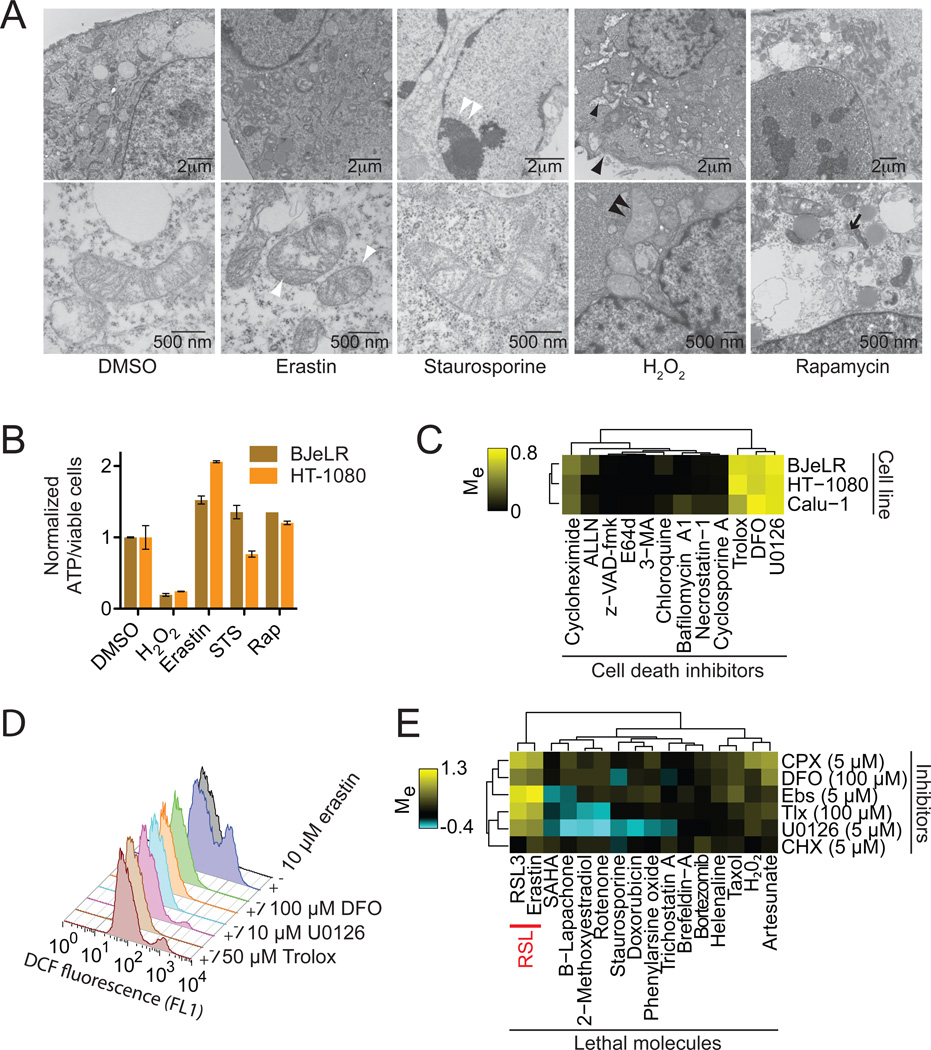
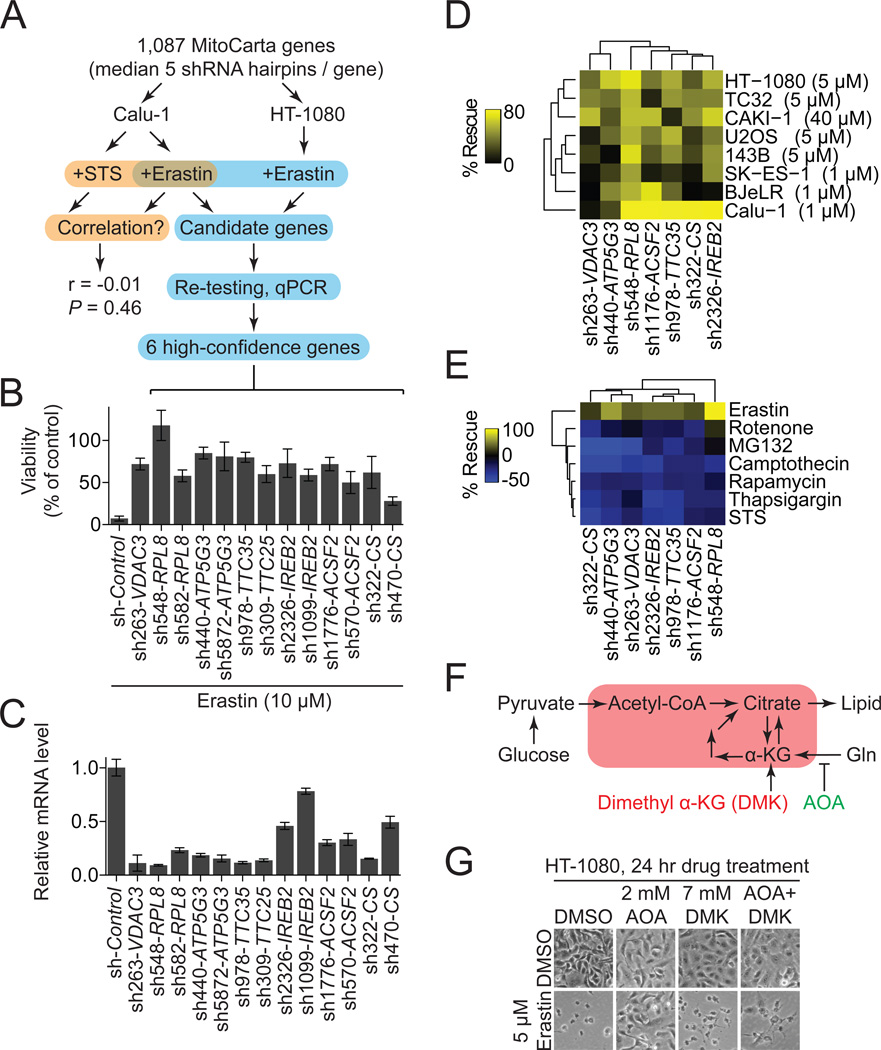
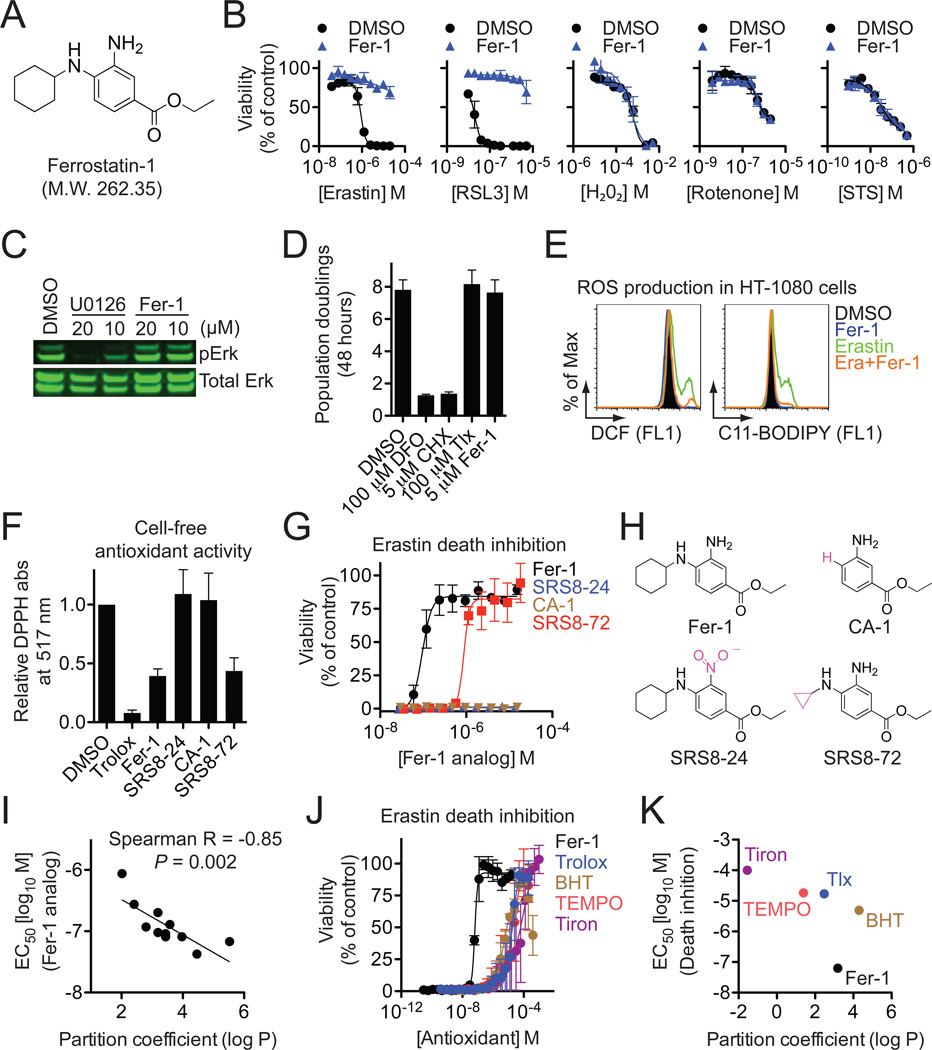
Comment in
-
Ironing out cell death mechanisms.Cell. 2012 May 25;149(5):963-5. doi: 10.1016/j.cell.2012.05.009. Cell. 2012. PMID: 22632964 No abstract available.
-
Ferroptosis: a novel cell death form will be a promising therapy target for diseases.Acta Biochim Biophys Sin (Shanghai). 2015 Oct;47(10):857-9. doi: 10.1093/abbs/gmv086. Epub 2015 Sep 7. Acta Biochim Biophys Sin (Shanghai). 2015. PMID: 26350095 No abstract available.
References
-
- Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, Kolle P, Tschoep K, Issels RD, Daniel PT, et al. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene. 2008;27:1618–1628. - PubMed
-
- Cater HL, Gitterman D, Davis SM, Benham CD, Morrison B, 3rd, Sundstrom LE. Stretch-induced injury in organotypic hippocampal slice cultures reproduces in vivo post-traumatic neurodegeneration: role of glutamate receptors and voltage-dependent calcium channels. J Neurochem. 2007;101:434–447. - PubMed
-
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
