

Durable pharmacological responses from the peptide ShK-186, a specific Kv1.3 channel inhibitor that suppresses T cell mediators of autoimmune disease
- PMID: 22637724
- PMCID: PMC3422530
- DOI: 10.1124/jpet.112.191890
Durable pharmacological responses from the peptide ShK-186, a specific Kv1.3 channel inhibitor that suppresses T cell mediators of autoimmune disease
Abstract
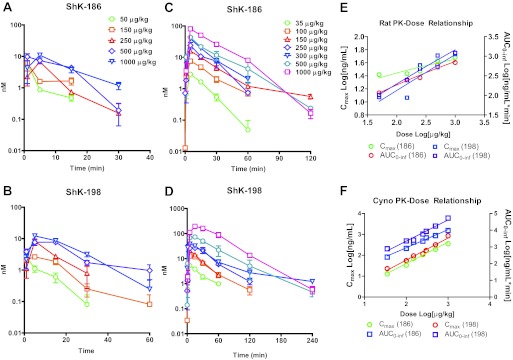
The Kv1.3 channel is a recognized target for pharmaceutical development to treat autoimmune diseases and organ rejection. ShK-186, a specific peptide inhibitor of Kv1.3, has shown promise in animal models of multiple sclerosis and rheumatoid arthritis. Here, we describe the pharmacokinetic-pharmacodynamic relationship for ShK-186 in rats and monkeys. The pharmacokinetic profile of ShK-186 was evaluated with a validated high-performance liquid chromatography-tandem mass spectrometry method to measure the peptide's concentration in plasma. These results were compared with single-photon emission computed tomography/computed tomography data collected with an ¹¹¹In-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-conjugate of ShK-186 to assess whole-blood pharmacokinetic parameters as well as the peptide's absorption, distribution, and excretion. Analysis of these data support a model wherein ShK-186 is absorbed slowly from the injection site, resulting in blood concentrations above the Kv1.3 channel-blocking IC₅₀ value for up to 7 days in monkeys. Pharmacodynamic studies on human peripheral blood mononuclear cells showed that brief exposure to ShK-186 resulted in sustained suppression of cytokine responses and may contribute to prolonged drug effects. In delayed-type hypersensitivity, chronic relapsing-remitting experimental autoimmune encephalomyelitis, and pristane-induced arthritis rat models, a single dose of ShK-186 every 2 to 5 days was as effective as daily administration. ShK-186's slow distribution from the injection site and its long residence time on the Kv1.3 channel contribute to the prolonged therapeutic effect of ShK-186 in animal models of autoimmune disease.
Figures






References
-
- Audebert F, Urtizberea M, Sabouraud A, Scherrmann JM, Bon C. (1994) Pharmacokinetics of Vipera aspis venom after experimental envenomation in rabbits. J Pharmacol Exp Ther 268:1512–1517 - PubMed
-
- Barral-Netto M, Schriefer A, Vinhas V, Almeida AR. (1990) Enzyme-linked immunosorbent assay for the detection of Bothrops jararaca venom. Toxicon 28:1053–1061 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
