

Validation of macromolecular flexibility in solution by small-angle X-ray scattering (SAXS)
- PMID: 22639100
- PMCID: PMC3462898
- DOI: 10.1007/s00249-012-0820-x
Validation of macromolecular flexibility in solution by small-angle X-ray scattering (SAXS)
Abstract
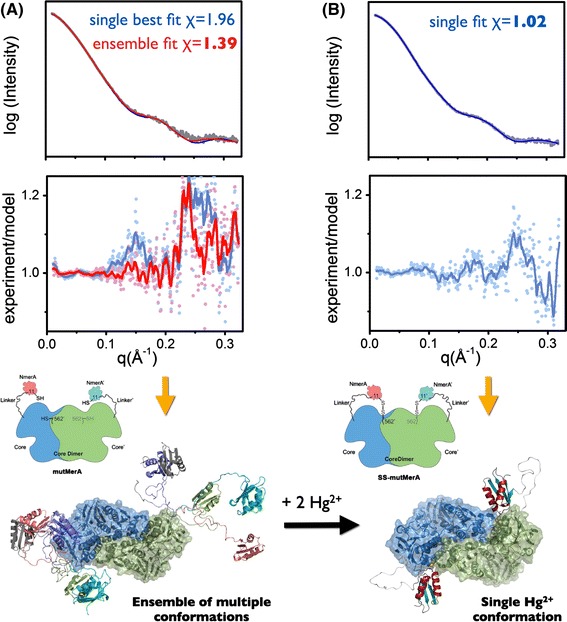
The dynamics of macromolecular conformations are critical to the action of cellular networks. Solution X-ray scattering studies, in combination with macromolecular X-ray crystallography (MX) and nuclear magnetic resonance (NMR), strive to determine complete and accurate states of macromolecules, providing novel insights describing allosteric mechanisms, supramolecular complexes, and dynamic molecular machines. This review addresses theoretical and practical concepts, concerns, and considerations for using these techniques in conjunction with computational methods to productively combine solution-scattering data with high-resolution structures. I discuss the principal means of direct identification of macromolecular flexibility from SAXS data followed by critical concerns about the methods used to calculate theoretical SAXS profiles from high-resolution structures. The SAXS profile is a direct interrogation of the thermodynamic ensemble and techniques such as, for example, minimal ensemble search (MES), enhance interpretation of SAXS experiments by describing the SAXS profiles as population-weighted thermodynamic ensembles. I discuss recent developments in computational techniques used for conformational sampling, and how these techniques provide a basis for assessing the level of the flexibility within a sample. Although these approaches sacrifice atomic detail, the knowledge gained from ensemble analysis is often appropriate for developing hypotheses and guiding biochemical experiments. Examples of the use of SAXS and combined approaches with X-ray crystallography, NMR, and computational methods to characterize dynamic assemblies are presented.
Figures
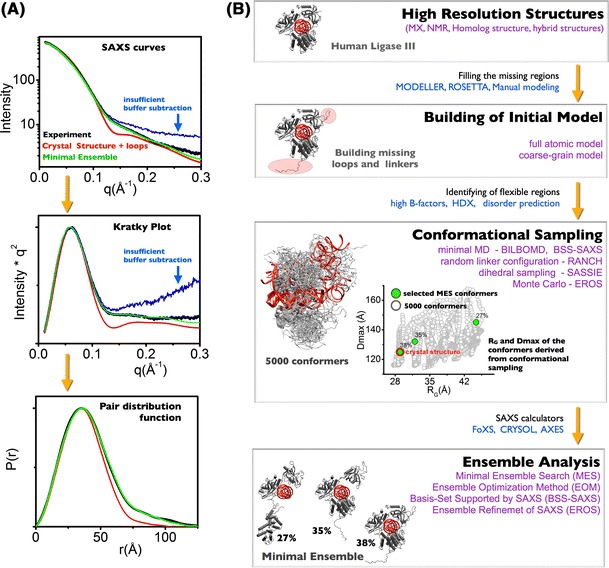
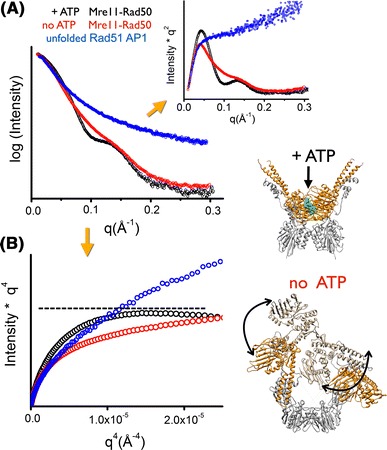
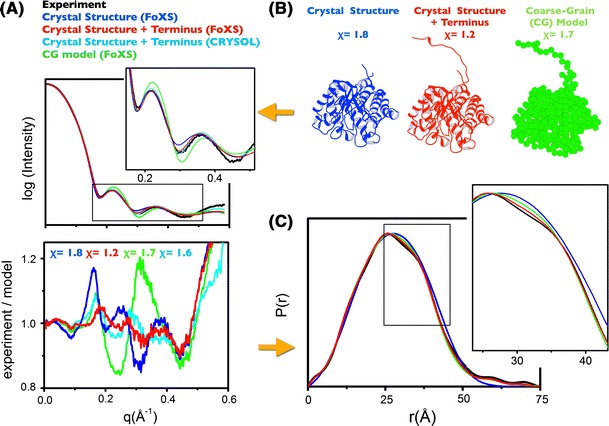
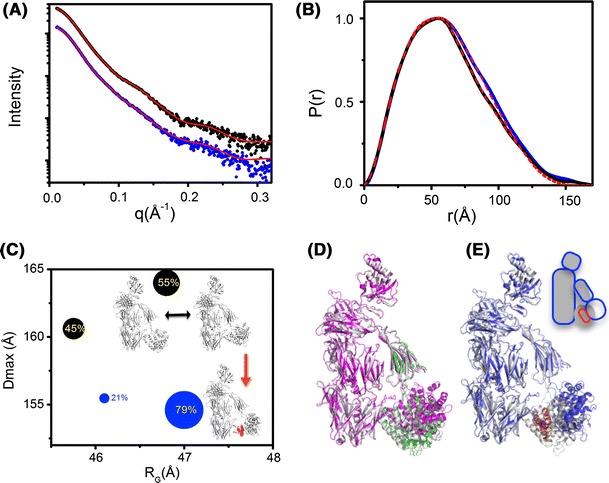
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
