

Leaky ryanodine receptors in β-sarcoglycan deficient mice: a potential common defect in muscular dystrophy
- PMID: 22640601
- PMCID: PMC3605002
- DOI: 10.1186/2044-5040-2-9
Leaky ryanodine receptors in β-sarcoglycan deficient mice: a potential common defect in muscular dystrophy
Abstract
Background: Disruption of the sarcolemma-associated dystrophin-glycoprotein complex underlies multiple forms of muscular dystrophy, including Duchenne muscular dystrophy and sarcoglycanopathies. A hallmark of these disorders is muscle weakness. In a murine model of Duchenne muscular dystrophy, mdx mice, cysteine-nitrosylation of the calcium release channel/ryanodine receptor type 1 (RyR1) on the skeletal muscle sarcoplasmic reticulum causes depletion of the stabilizing subunit calstabin1 (FKBP12) from the RyR1 macromolecular complex. This results in a sarcoplasmic reticular calcium leak via defective RyR1 channels. This pathological intracellular calcium leak contributes to reduced calcium release and decreased muscle force production. It is unknown whether RyR1 dysfunction occurs also in other muscular dystrophies.
Methods: To test this we used a murine model of Limb-Girdle muscular dystrophy, deficient in β-sarcoglycan (Sgcb-/-).
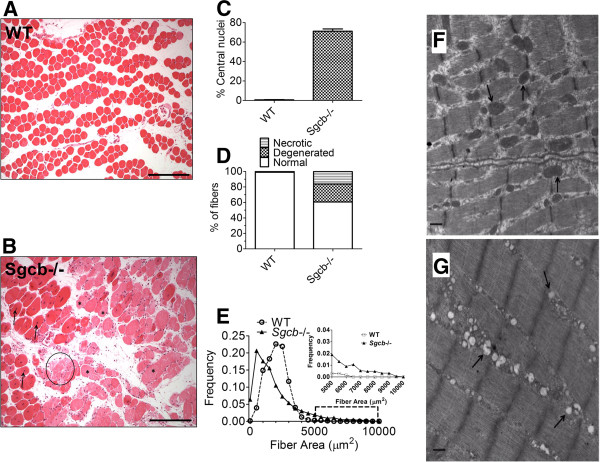
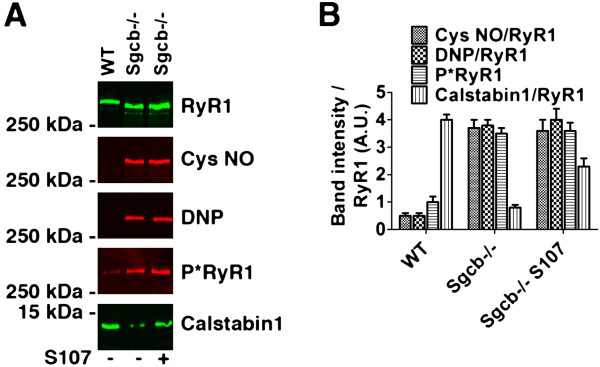
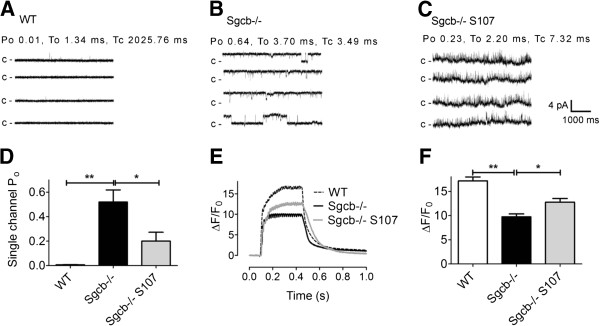
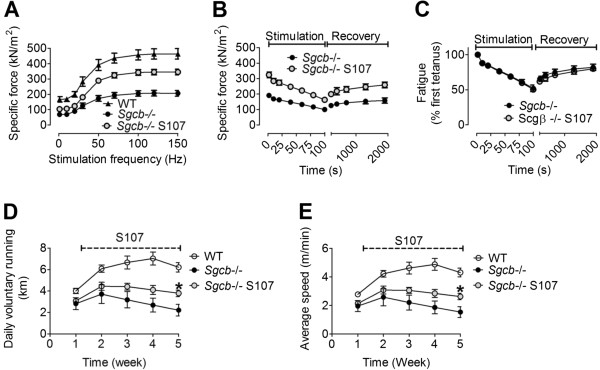
Results: Skeletal muscle RyR1 from Sgcb-/- deficient mice were oxidized, nitrosylated, and depleted of the stabilizing subunit calstabin1, which was associated with increased open probability of the RyR1 channels. Sgcb-/- deficient mice exhibited decreased muscle specific force and calcium transients, and displayed reduced exercise capacity. Treating Sgcb-/- mice with the RyR stabilizing compound S107 improved muscle specific force, calcium transients, and exercise capacity. We have previously reported similar findings in mdx mice, a murine model of Duchenne muscular dystrophy.
Conclusions: Our data suggest that leaky RyR1 channels may underlie multiple forms of muscular dystrophy linked to mutations in genes encoding components of the dystrophin-glycoprotein complex. A common underlying abnormality in calcium handling indicates that pharmacological targeting of dysfunctional RyR1 could be a novel therapeutic approach to improve muscle function in Limb-Girdle and Duchenne muscular dystrophies.
Figures
References
-
- Lim LE, Duclos F, Broux O, Bourg N, Sunada Y, Allamand V, Meyer J, Richard I, Moomaw C, Slaughter C, Tome FMS, Fardeau M, Jackson CE, Beckmann JS, Campbell KP. Beta-sarcoglycan: characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet. 1995;11:257–265. doi: 10.1038/ng1195-257. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
