

Prevention of methamphetamine-induced microglial cell death by TNF-α and IL-6 through activation of the JAK-STAT pathway
- PMID: 22642790
- PMCID: PMC3391183
- DOI: 10.1186/1742-2094-9-103
Prevention of methamphetamine-induced microglial cell death by TNF-α and IL-6 through activation of the JAK-STAT pathway
Abstract
Background: It is well known that methamphetamine (METH) is neurotoxic and recent studies have suggested the involvement of neuroinflammatory processes in brain dysfunction induced by misuse of this drug. Indeed, glial cells seem to be activated in response to METH, but its effects on microglial cells are not fully understood. Moreover, it has been shown that cytokines, which are normally released by activated microglia, may have a dual role in response to brain injury. This led us to study the toxic effect of METH on microglial cells by looking to cell death and alterations of tumor necrosis factor-alpha (TNF-α) and interleukine-6 (IL-6) systems, as well as the role played by these cytokines.
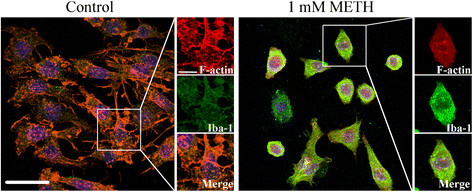
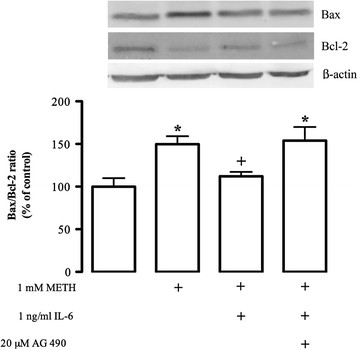
Methods: We used the N9 microglial cell line, and cell death and proliferation were evaluated by terminal deoxynucleotidyl transferase dUTP nick end labeling assay and incorporation of bromodeoxyuridine, respectively. The TNF-α and IL-6 content was quantified by enzyme-linked immunosorbent assay, and changes in TNF receptor 1, IL-6 receptor-alpha, Bax and Bcl-2 protein levels by western blotting. Immunocytochemistry analysis was also performed to evaluate alterations in microglial morphology and in the protein expression of phospho-signal transducer and activator of transcription 3 (pSTAT3).
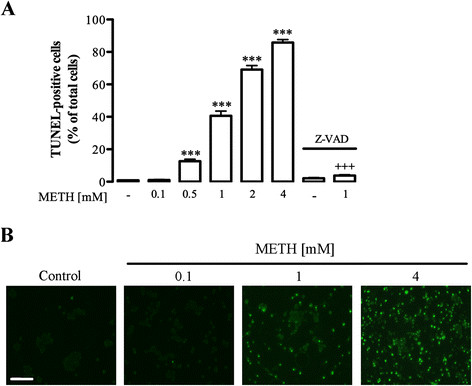
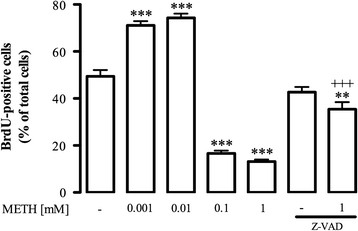
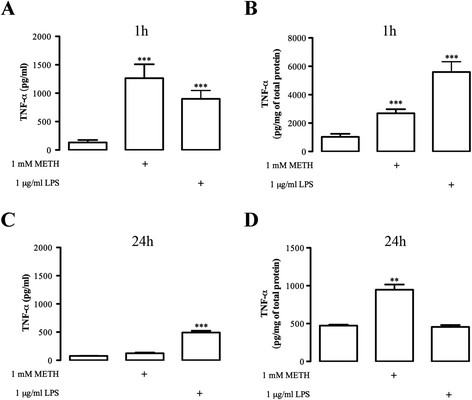
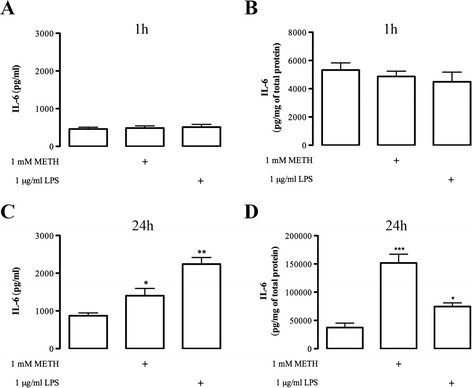
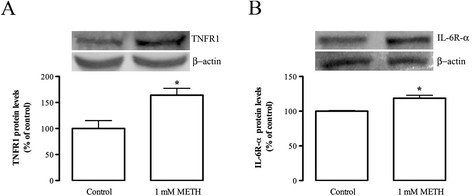
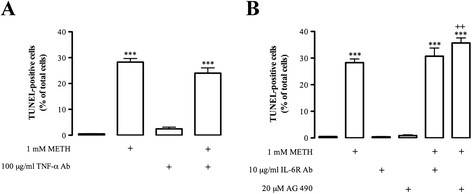
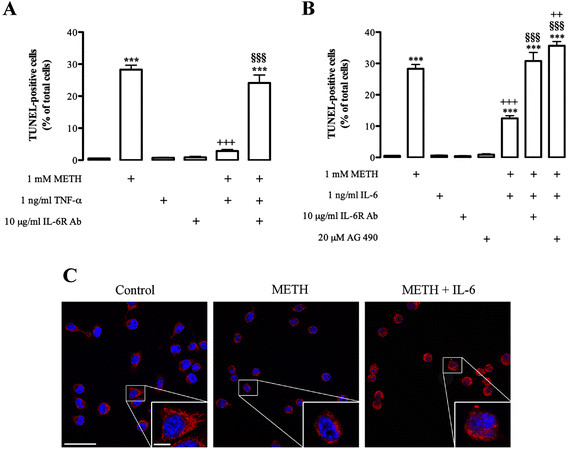
Results: METH induced microglial cell death in a concentration-dependent manner (EC50 = 1 mM), and also led to significant morphological changes and decreased cell proliferation. Additionally, this drug increased TNF-α extracellular and intracellular levels, as well as its receptor protein levels at 1 h, whereas IL-6 and its receptor levels were increased at 24 h post-exposure. However, the endogenous proinflammatory cytokines did not contribute to METH-induced microglial cell death. On the other hand, exogenous low concentrations of TNF-α or IL-6 had a protective effect. Interestingly, we also verified that the anti-apoptotic role of TNF-α was mediated by activation of IL-6 signaling, specifically the janus kinase (JAK)-STAT3 pathway, which in turn induced down-regulation of the Bax/Bcl-2 ratio.
Conclusions: These findings show that TNF-α and IL-6 have a protective role against METH-induced microglial cell death via the IL-6 receptor, specifically through activation of the JAK-STAT3 pathway, with consequent changes in pro- and anti-apoptotic proteins.
Figures
Similar articles
-
Methamphetamine-induced cardiotoxicity: in search of protective transcriptional mechanisms.Herz. 2024 Dec;49(6):434-440. doi: 10.1007/s00059-024-05279-6. Epub 2024 Oct 25. Herz. 2024. PMID: 39455447 Free PMC article. Review.
-
Asiatic acid attenuates methamphetamine-induced neuroinflammation and neurotoxicity through blocking of NF-kB/STAT3/ERK and mitochondria-mediated apoptosis pathway.J Neuroinflammation. 2017 Dec 11;14(1):240. doi: 10.1186/s12974-017-1009-0. J Neuroinflammation. 2017. PMID: 29228978 Free PMC article.
-
The role of the JAK2-STAT3 pathway in pro-inflammatory responses of EMF-stimulated N9 microglial cells.J Neuroinflammation. 2010 Sep 9;7:54. doi: 10.1186/1742-2094-7-54. J Neuroinflammation. 2010. PMID: 20828402 Free PMC article.
-
Methamphetamine modulates the production of interleukin-6 and tumor necrosis factor-alpha via the cAMP/PKA/CREB signaling pathway in lipopolysaccharide-activated microglia.Int Immunopharmacol. 2018 Mar;56:168-178. doi: 10.1016/j.intimp.2018.01.024. Epub 2018 Feb 3. Int Immunopharmacol. 2018. PMID: 29414647
-
The Role of IL-6 in Fibrotic Diseases: Molecular and Cellular Mechanisms.Int J Biol Sci. 2022 Aug 29;18(14):5405-5414. doi: 10.7150/ijbs.75876. eCollection 2022. Int J Biol Sci. 2022. PMID: 36147459 Free PMC article. Review.
Cited by
-
Melatonin Protects Methamphetamine-Induced Neuroinflammation Through NF-κB and Nrf2 Pathways in Glioma Cell Line.Neurochem Res. 2015 Jul;40(7):1448-56. doi: 10.1007/s11064-015-1613-2. Epub 2015 May 22. Neurochem Res. 2015. PMID: 25998888
-
Methamphetamine alters microglial immune function through P2X7R signaling.J Neuroinflammation. 2016 Apr 26;13(1):91. doi: 10.1186/s12974-016-0553-3. J Neuroinflammation. 2016. PMID: 27117066 Free PMC article.
-
Lupenone Protects Neuroblastoma SH-SY5y Cells Against Methamphetamine-Induced Apoptotic Cell Death via PI3K/Akt/mTOR Signaling Pathway.Int J Mol Sci. 2020 Feb 27;21(5):1617. doi: 10.3390/ijms21051617. Int J Mol Sci. 2020. PMID: 32120831 Free PMC article.
-
Methamphetamine-induced cardiotoxicity: in search of protective transcriptional mechanisms.Herz. 2024 Dec;49(6):434-440. doi: 10.1007/s00059-024-05279-6. Epub 2024 Oct 25. Herz. 2024. PMID: 39455447 Free PMC article. Review.
-
Microglia contribute to methamphetamine reinforcement and reflect persistent transcriptional and morphological adaptations to the drug.bioRxiv [Preprint]. 2024 Feb 17:2023.10.19.563168. doi: 10.1101/2023.10.19.563168. bioRxiv. 2024. Update in: Brain Behav Immun. 2024 Aug;120:339-351. doi: 10.1016/j.bbi.2024.05.038. PMID: 37961443 Free PMC article. Updated. Preprint.
References
-
- Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K, Iwata Y, Tsuchiya KJ, Suda S, Suzuki K, Kawai M, Takebayashi K, Yamamoto S, Matsuzaki H, Ueki T, Mori N, Gold MS, Cadet JL. Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci. 2008;28:5756–5761. doi: 10.1523/JNEUROSCI.1179-08.2008. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
