

Suggested guidelines for the diagnosis and management of urea cycle disorders
- PMID: 22642880
- PMCID: PMC3488504
- DOI: 10.1186/1750-1172-7-32
Suggested guidelines for the diagnosis and management of urea cycle disorders
Abstract
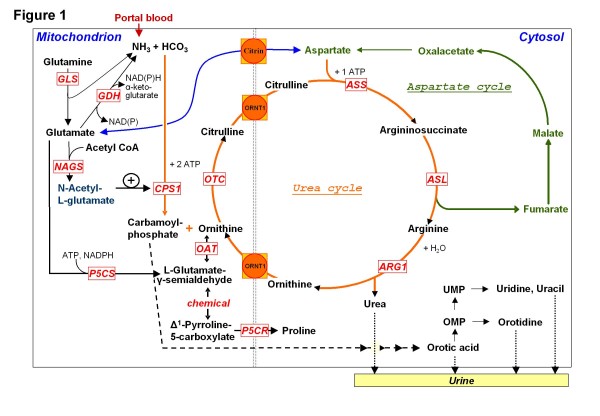
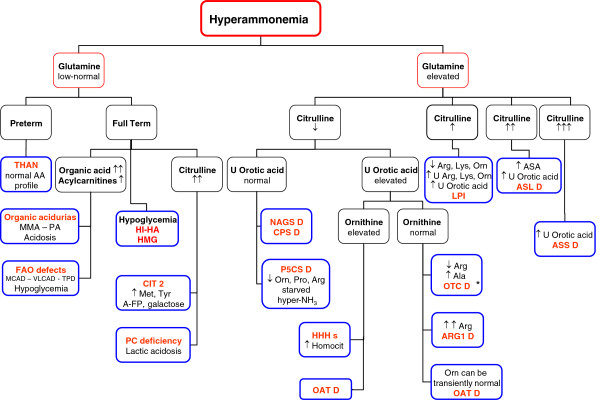
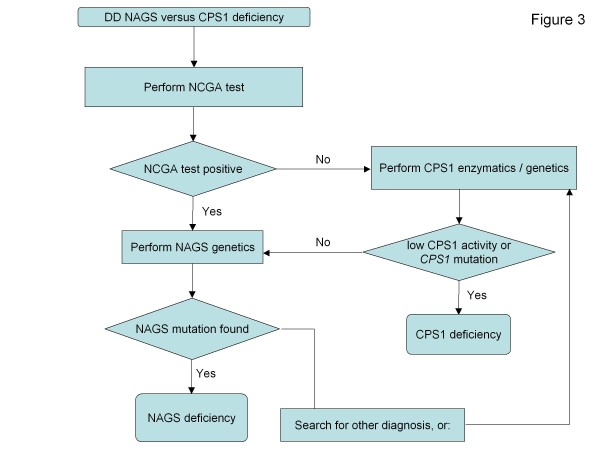
Urea cycle disorders (UCDs) are inborn errors of ammonia detoxification/arginine synthesis due to defects affecting the catalysts of the Krebs-Henseleit cycle (five core enzymes, one activating enzyme and one mitochondrial ornithine/citrulline antiporter) with an estimated incidence of 1:8.000. Patients present with hyperammonemia either shortly after birth (~50%) or, later at any age, leading to death or to severe neurological handicap in many survivors. Despite the existence of effective therapy with alternative pathway therapy and liver transplantation, outcomes remain poor. This may be related to underrecognition and delayed diagnosis due to the nonspecific clinical presentation and insufficient awareness of health care professionals because of disease rarity. These guidelines aim at providing a trans-European consensus to: guide practitioners, set standards of care and help awareness campaigns. To achieve these goals, the guidelines were developed using a Delphi methodology, by having professionals on UCDs across seven European countries to gather all the existing evidence, score it according to the SIGN evidence level system and draw a series of statements supported by an associated level of evidence. The guidelines were revised by external specialist consultants, unrelated authorities in the field of UCDs and practicing pediatricians in training. Although the evidence degree did hardly ever exceed level C (evidence from non-analytical studies like case reports and series), it was sufficient to guide practice on both acute and chronic presentations, address diagnosis, management, monitoring, outcomes, and psychosocial and ethical issues. Also, it identified knowledge voids that must be filled by future research. We believe these guidelines will help to: harmonise practice, set common standards and spread good practices with a positive impact on the outcomes of UCD patients.
Figures
References
-
- Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127–170. - PubMed
-
- Dionisi-Vici C, Rizzo C, Burlina AB, Caruso U, Sabetta G, Uziel G, Abeni D. Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey. J Pediatr. 2002;140:321–327. - PubMed
-
- Wilcken B. Problems in the management of urea cycle disorders. Mol Genet Metab. 2004;81(Suppl 1):S86–S91. - PubMed
-
- Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur J Pediatr. 2003;162:410–416. - PubMed
-
- Nassogne MC, Heron B, Touati G, Rabier D, Saudubray JM. Urea cycle defects: management and outcome. J Inherit Metab Dis. 2005;28:407–414. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
