

The use of modeling tools to drive efficient oral product design
- PMID: 22644702
- PMCID: PMC3385810
- DOI: 10.1208/s12248-012-9372-3
The use of modeling tools to drive efficient oral product design
Abstract
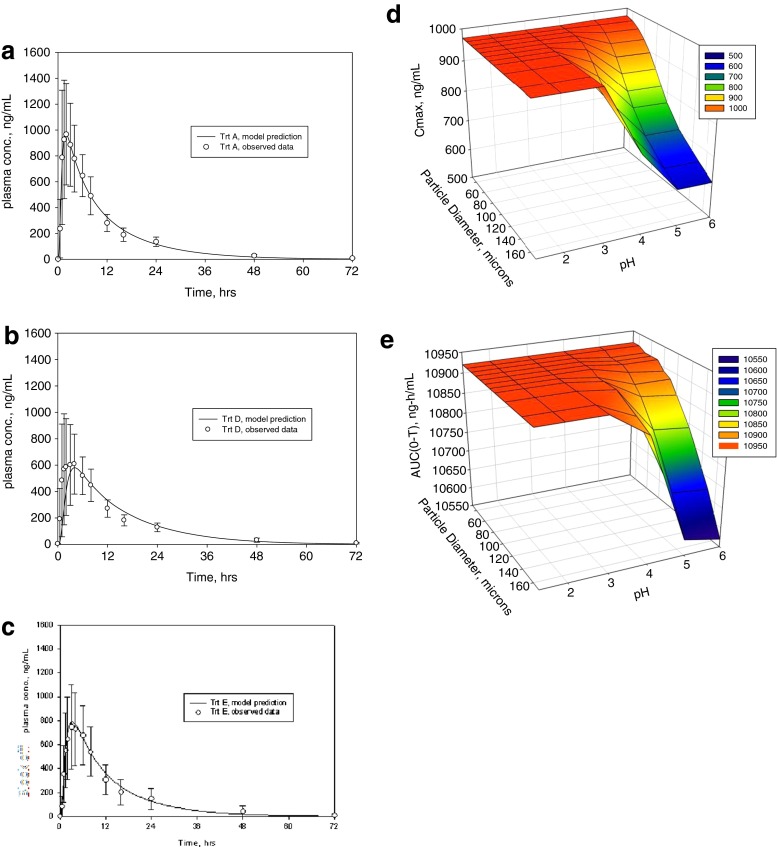
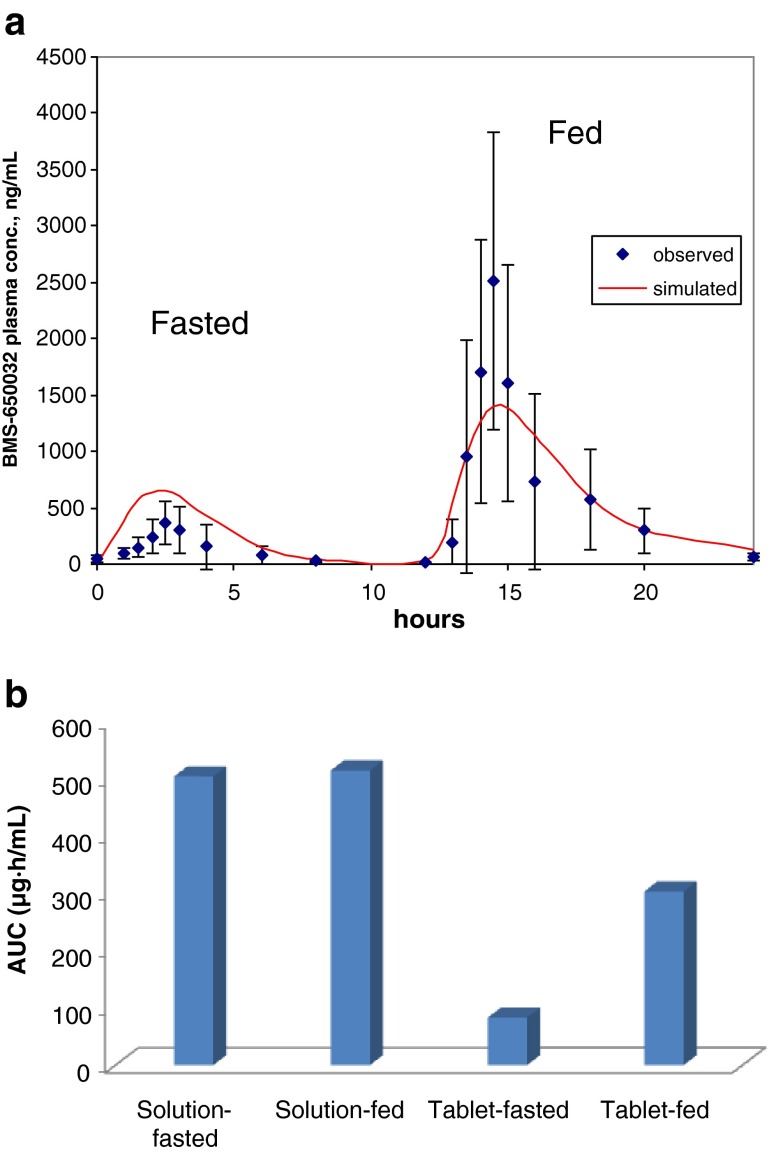
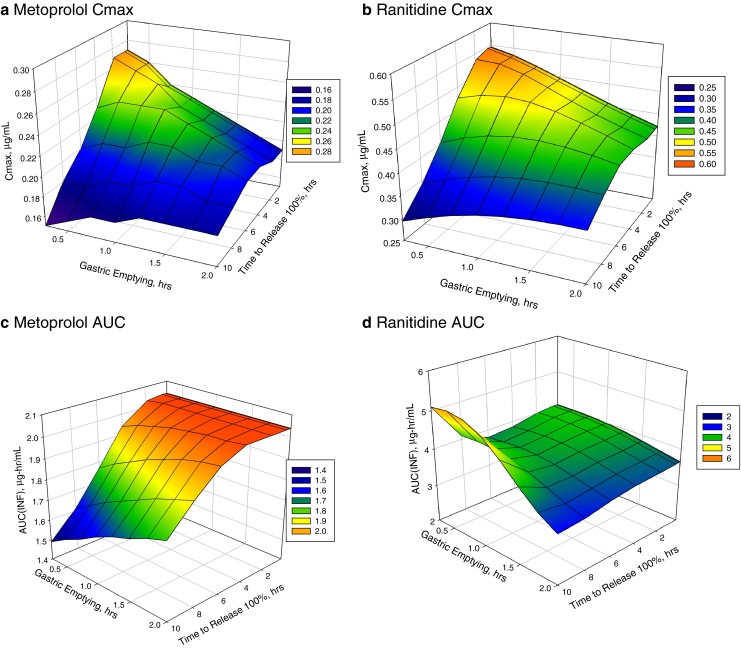
Modeling and simulation of drug dissolution and oral absorption has been increasingly used over the last decade to understand drug behavior in vivo based on the physicochemical properties of Active Pharmaceutical Ingredients (API) and dosage forms. As in silico and in vitro tools become more sophisticated and our knowledge of physiological processes has grown, model simulations can provide a valuable confluence, tying-in in vitro data with in vivo data while offering mechanistic insights into clinical performance. To a formulation scientist, this unveils not just the parameters that are predicted to significantly impact dissolution/absorption, but helps probe explanations around drug product performance and address specific in vivo mechanisms. In formulation, development, in silico dissolution-absorption modeling can be effectively used to guide: API selection (form comparison and particle size properties), influence clinical study design, assess dosage form performance, guide strategy for dosage form design, and breakdown clinically relevant conditions on dosage form performance (pH effect for patients on pH-elevating treatments, and food effect). This minireview describes examples of these applications in guiding product development including those with strategies to mitigate observed clinical exposure liability or mechanistically probe product in vivo performance attributes.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
