

Templates are available to model nearly all complexes of structurally characterized proteins
- PMID: 22645367
- PMCID: PMC3386081
- DOI: 10.1073/pnas.1200678109
Templates are available to model nearly all complexes of structurally characterized proteins
Abstract
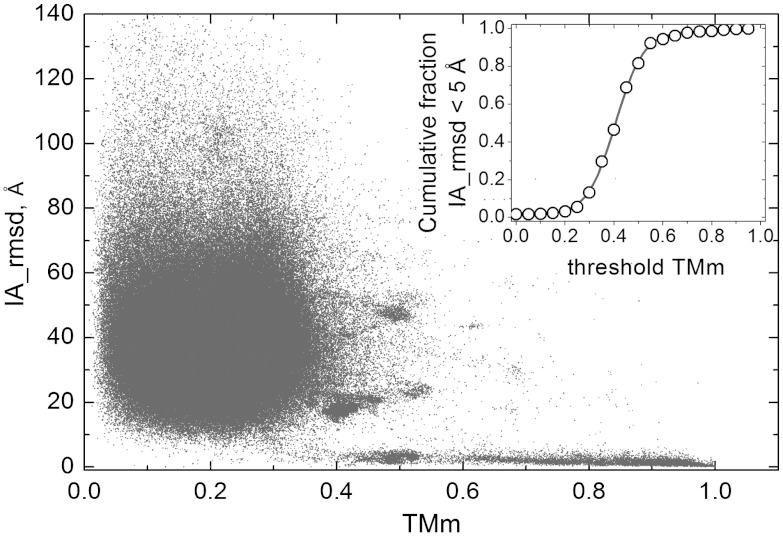
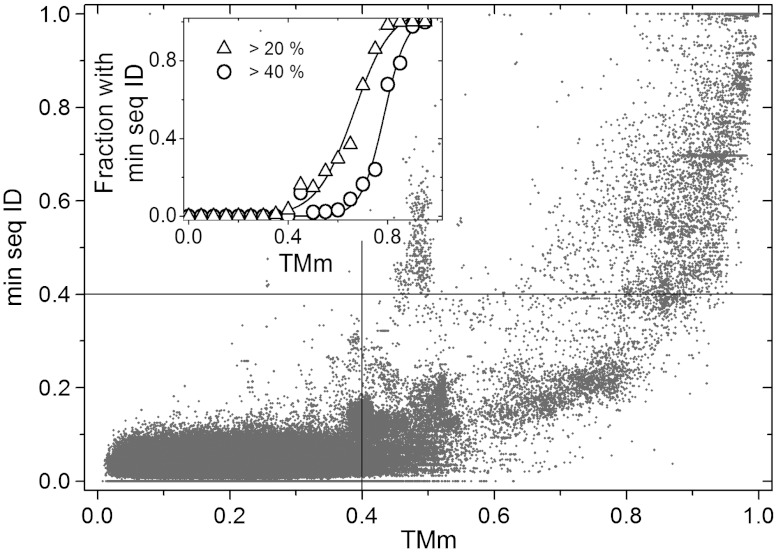
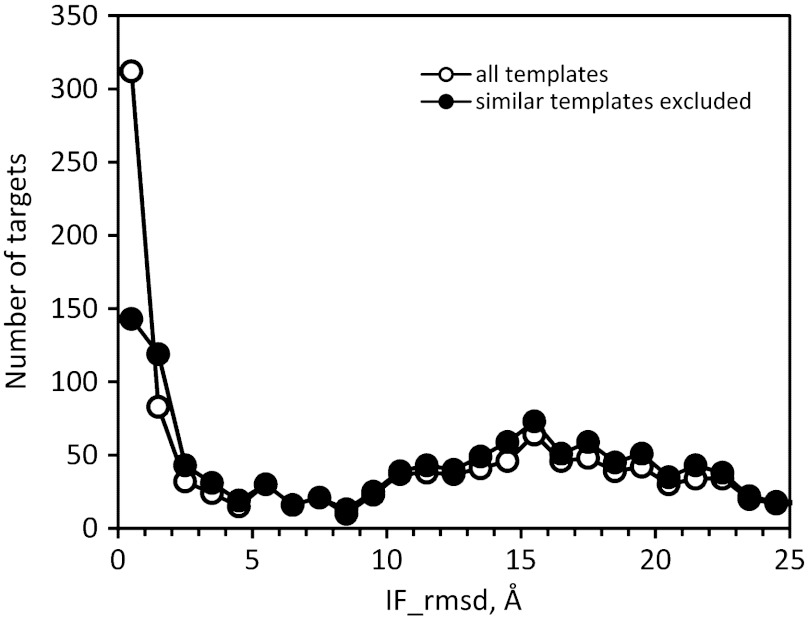
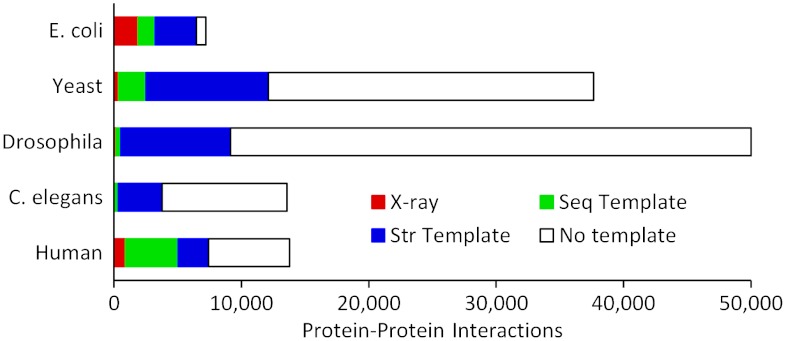
Traditional approaches to protein-protein docking sample the binding modes with no regard to similar experimentally determined structures (templates) of protein-protein complexes. Emerging template-based docking approaches utilize such similar complexes to determine the docking predictions. The docking problem assumes the knowledge of the participating proteins' structures. Thus, it provides the possibility of aligning the structures of the proteins and the template complexes. The progress in the development of template-based docking and the vast experience in template-based modeling of individual proteins show that, generally, such approaches are more reliable than the free modeling. The key aspect of this modeling paradigm is the availability of the templates. The current common perception is that due to the difficulties in experimental structure determination of protein-protein complexes, the pool of docking templates is insignificant, and thus a broad application of template-based docking is possible only at some future time. The results of our large scale, systematic study show that, surprisingly, in spite of the limited number of protein-protein complexes in the Protein Data Bank, docking templates can be found for complexes representing almost all the known protein-protein interactions, provided the components themselves have a known structure or can be homology-built. About one-third of the templates are of good quality when they are compared to experimental structures in test sets extracted from the Protein Data Bank and would be useful starting points in modeling the complexes. This finding dramatically expands our ability to model protein interactions, and has far-reaching implications for the protein docking field in general.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Janin J, Bahadur RP, Chakrabarti P. Protein–protein interaction and quaternary structure. Q Rev Biophys. 2008;41:133–180. - PubMed
-
- Kozakov D, Brenke R, Comeau SR, Vajda S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. - PubMed
-
- Mintseris J, et al. Integrating statistical pair potentials into protein complex prediction. Proteins. 2007;69:511–520. - PubMed
-
- de Vries SJ, et al. HADDOCK versus HADDOCK: New features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
