

Alzheimer's disease, β-amyloid, glutamate, NMDA receptors and memantine--searching for the connections
- PMID: 22646481
- PMCID: PMC3481041
- DOI: 10.1111/j.1476-5381.2012.02057.x
Alzheimer's disease, β-amyloid, glutamate, NMDA receptors and memantine--searching for the connections
Abstract
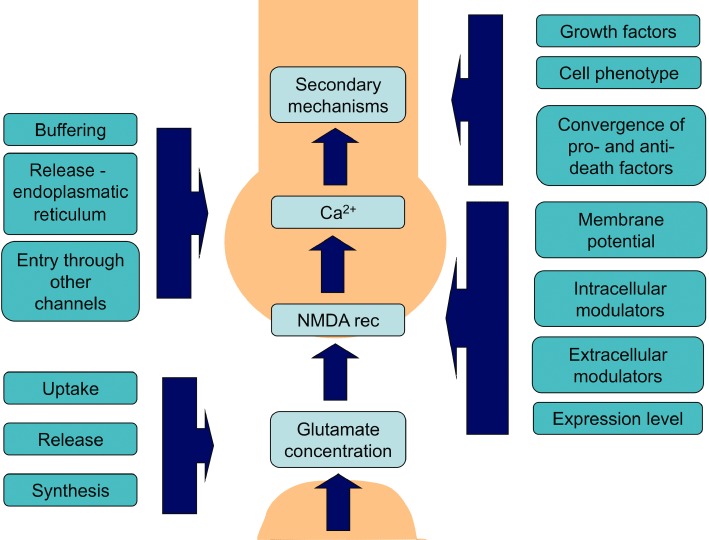
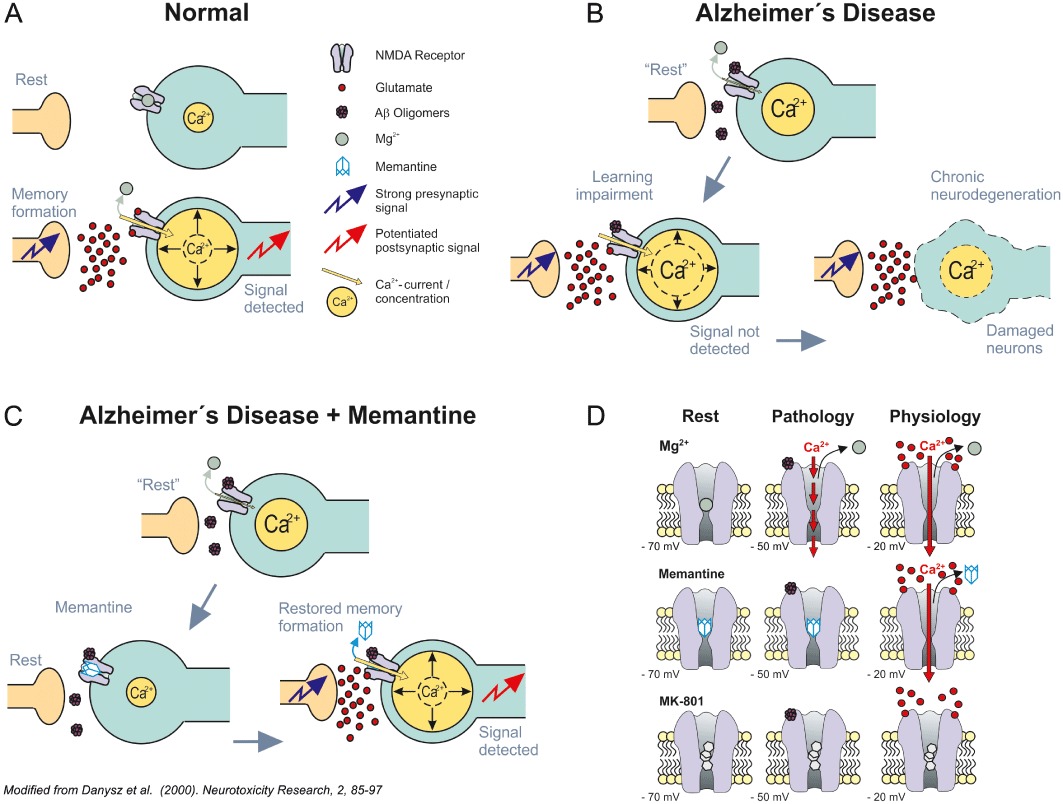
β-amyloid (Aβ) is widely accepted to be one of the major pathomechanisms underlying Alzheimer's disease (AD), although there is presently lively debate regarding the relative roles of particular species/forms of this peptide. Most recent evidence indicates that soluble oligomers rather than plaques are the major cause of synaptic dysfunction and ultimately neurodegeneration. Soluble oligomeric Aβ has been shown to interact with several proteins, for example glutamatergic receptors of the NMDA type and proteins responsible for maintaining glutamate homeostasis such as uptake and release. As NMDA receptors are critically involved in neuronal plasticity including learning and memory, we felt that it would be valuable to provide an up to date review of the evidence connecting Aβ to these receptors and related neuronal plasticity. Strong support for the clinical relevance of such interactions is provided by the NMDA receptor antagonist memantine. This substance is the only NMDA receptor antagonist used clinically in the treatment of AD and therefore offers an excellent tool to facilitate translational extrapolations from in vitro studies through in vivo animal experiments to its ultimate clinical utility.
© 2012 The Authors. British Journal of Pharmacology © 2012 The British Pharmacological Society.
Figures
References
-
- Abbott JJ, Howlett DR, Francis PT, Williams RJ. Abeta(1-42) modulation of Akt phosphorylation via alpha7 nAChR and NMDA receptors. Neurobiol Aging. 2007;29:992–1001. - PubMed
-
- Abe K, Misawa M. Amyloid beta protein enhances the clearance of extracellular L-glutamate by cultured rat cortical astrocytes. Neurosci Res. 2003;45:25–31. - PubMed
-
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. - PubMed
-
- Alberdi E, Sanchez-Gomez MV, Cavaliere F, Perez-Samartin A, Zugaza JL, Trullas R, et al. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium. 2010;47:264–272. - PubMed
-
- Albrecht M, Rammes G, Parsons CG. 2008. Memantine reverses β-amyloid oligomers-induced deficits in long term potentiation (LTP) in murine hippocampal slices. 38th Society for Neuroscience Annual Meeting. Washington, USA. 15-11-2008. Society for Neuroscience Abstracts. 34, #829.21.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
