

KCa 3.1 channels maintain endothelium-dependent vasodilatation in isolated perfused kidneys of spontaneously hypertensive rats after chronic inhibition of NOS
- PMID: 22646737
- PMCID: PMC3575784
- DOI: 10.1111/j.1476-5381.2012.02062.x
KCa 3.1 channels maintain endothelium-dependent vasodilatation in isolated perfused kidneys of spontaneously hypertensive rats after chronic inhibition of NOS
Abstract
Background and purpose: The purpose of the study was to investigate renal endothelium-dependent vasodilatation in a model of severe hypertension associated with kidney injury.
Experimental approach: Changes in perfusion pressure were measured in isolated, perfused kidneys taken from 18-week-old Wistar-Kyoto rat (WKY), spontaneously hypertensive rats (SHR) and SHR treated for 2 weeks with N(ω) -nitro-L-arginine methyl ester in the drinking water (L-NAME-treated SHR, 6 mg·kg(-1) ·day(-1) ).
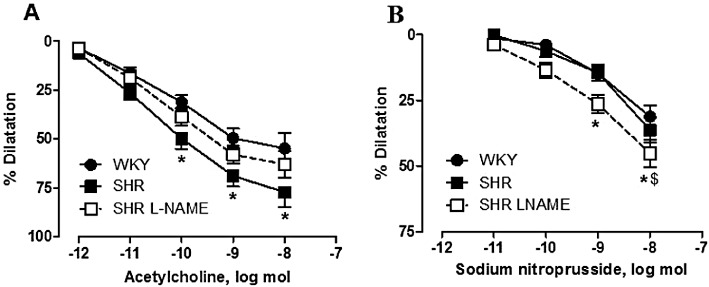
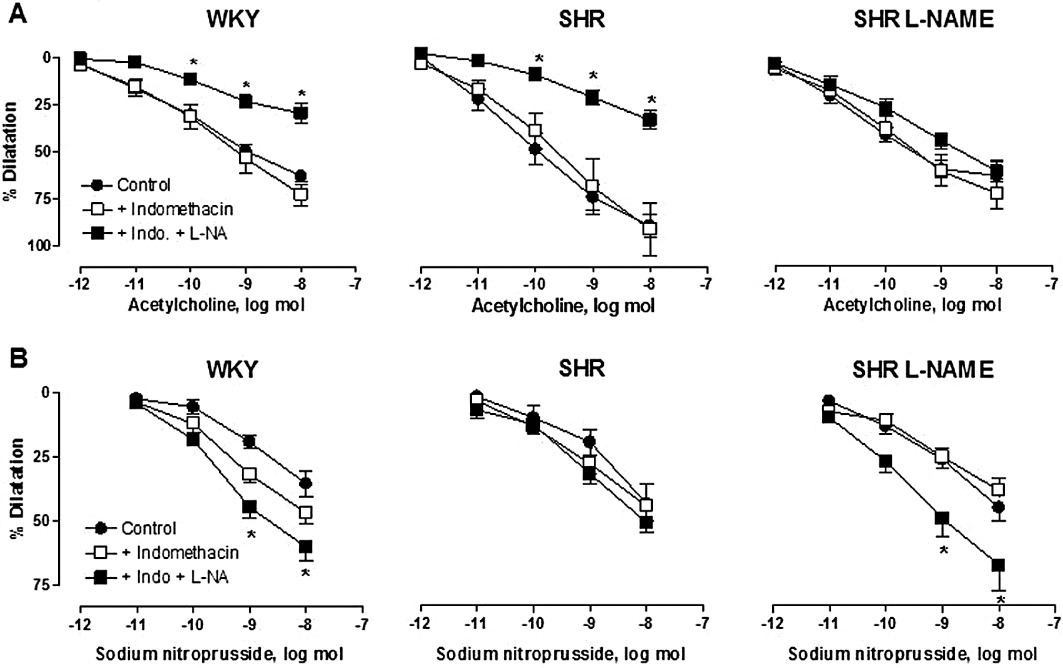
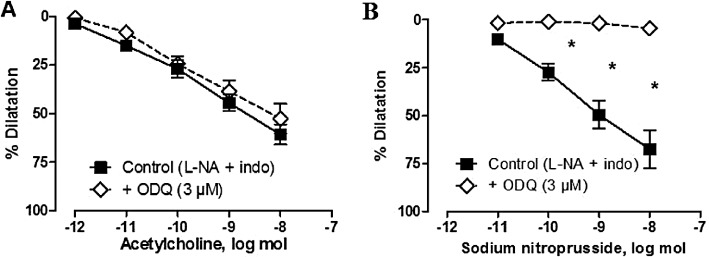
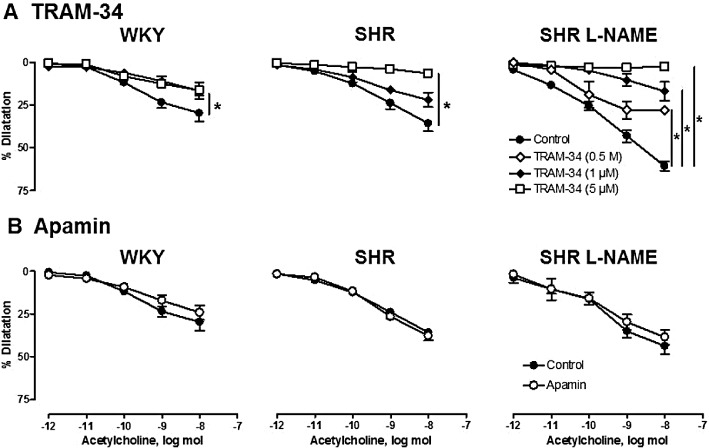
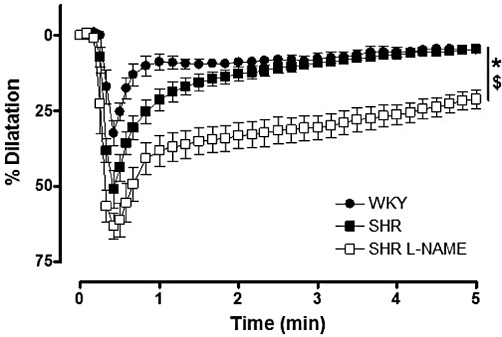
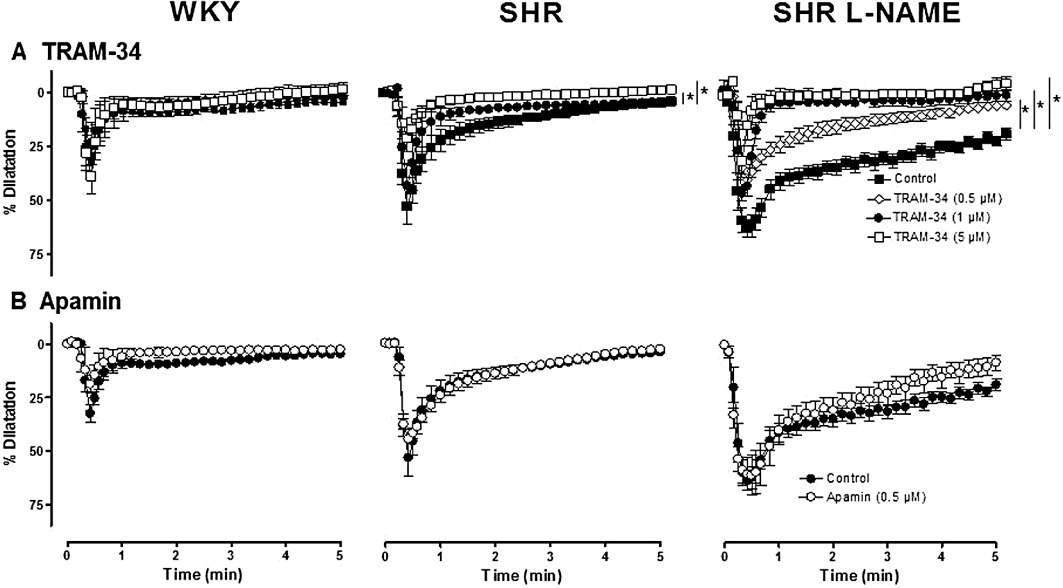
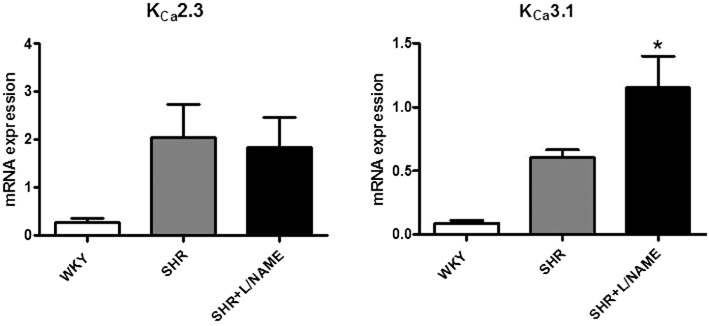
Key results: Acetylcholine caused similar dose-dependent renal dilatation in the three groups. In vitro administration of indomethacin did not alter the vasodilatation, while the addition of N(w) -nitro-L-arginine (L-NA) produced a differential inhibition of the vasodilatation, (inhibition in WKY > SHR > L-NAME-treated SHR). Further addition of ODQ, an inhibitor of soluble guanylyl cyclase, abolished the responses to sodium nitroprusside but did not affect the vasodilatation to acetylcholine. However, the addition of TRAM-34 (or charybdotoxin) inhibitors of Ca(2+) -activated K(+) channels of intermediate conductance (K(Ca) 3.1), blocked the vasodilatation to acetylcholine, while apamin, an inhibitor of Ca(2+) -activated K(+) channels of small conductance (K(Ca) 2.3), was ineffective. Dilatation induced by an opener of K(Ca) 3.1/K(Ca) 2.3 channels, NS-309, was also blocked by TRAM-34, but not by apamin. The magnitude and duration of NS-309-induced vasodilatation and the renal expression of mRNA for K(Ca) 3.1, but not K(Ca) 2.3, channels followed the same ranking order (WKY < SHR < L-NAME-treated SHR).
Conclusions and implications: In SHR kidneys, an EDHF-mediated response, involving activation of K(Ca) 3.1 channels, contributed to the mechanism of endothelium-dependent vasodilatation. In kidneys from L-NAME-treated SHR, up-regulation of this pathway fully compensated for the decrease in NO availability.
© 2012 The Authors. British Journal of Pharmacology © 2012 The British Pharmacological Society.
Figures
References
-
- Baylis C, Qiu C. Importance of nitric oxide in the control of renal hemodynamics. Kidney Int. 1996;49:1727–1731. - PubMed
-
- Benter IF, Francis I, Cojocel C, Juggi JS, Yousif MH, Canatan H. Contribution of cytochrome metabolites of arachidonic acid to hypertension and end-organ damage in SHR treated with L-NAME. Auton Autacoid Pharmacol. 2005;25:143–154. - PubMed
-
- Brandes RP, Schmitz-Winnenthal F-H, Félétou M, Gödecke A, Huang P-L, Vanhoutte PM, et al. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild type and endothelial NO synthase knock-out mice. Proc Natl Acad Sci U S A. 2000;97:9747–9752. - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
