

Detection and correction of probe-level artefacts on microarrays
- PMID: 22647057
- PMCID: PMC3534149
- DOI: 10.1186/1471-2105-13-114
Detection and correction of probe-level artefacts on microarrays
Abstract
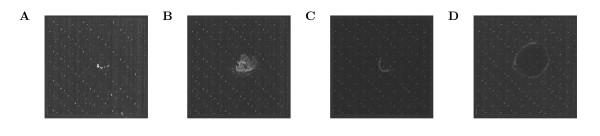
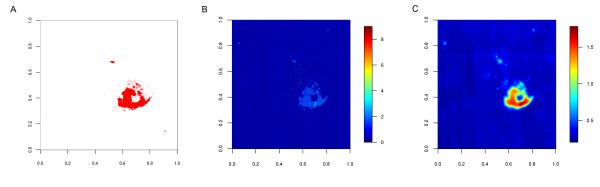
Background: A recent large-scale analysis of Gene Expression Omnibus (GEO) data found frequent evidence for spatial defects in a substantial fraction of Affymetrix microarrays in the GEO. Nevertheless, in contrast to quality assessment, artefact detection is not widely used in standard gene expression analysis pipelines. Furthermore, although approaches have been proposed to detect diverse types of spatial noise on arrays, the correction of these artefacts is mostly left to either summarization methods or the corresponding arrays are completely discarded.
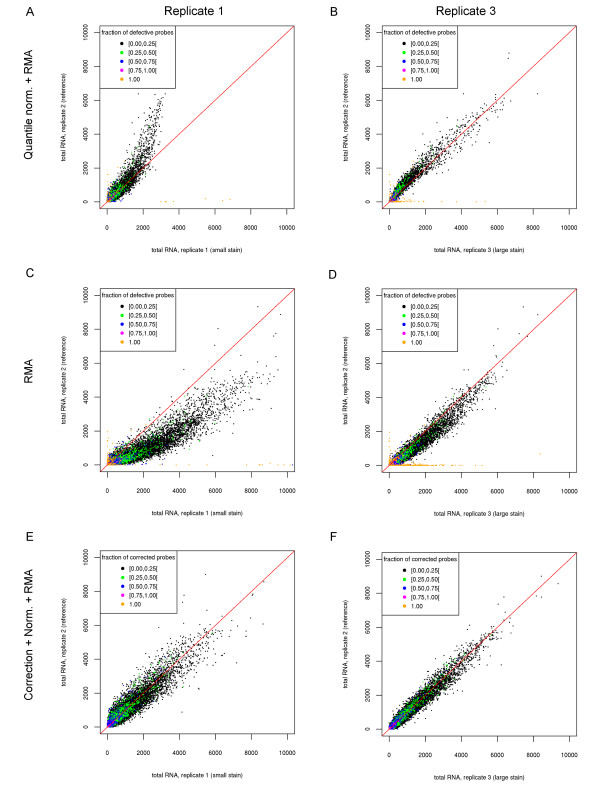
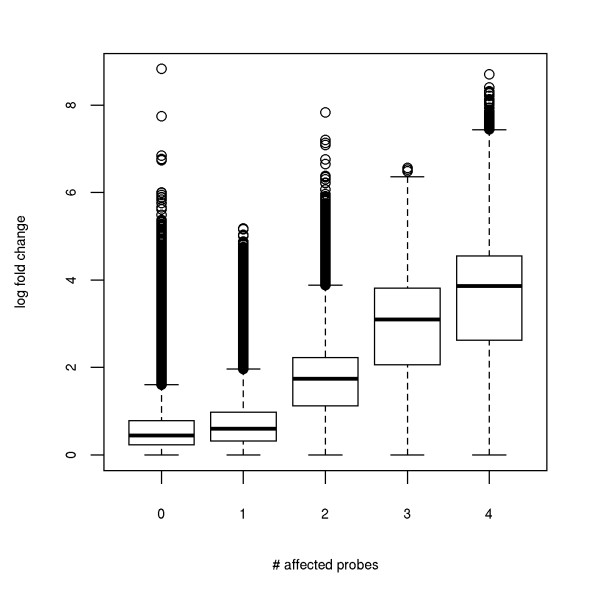
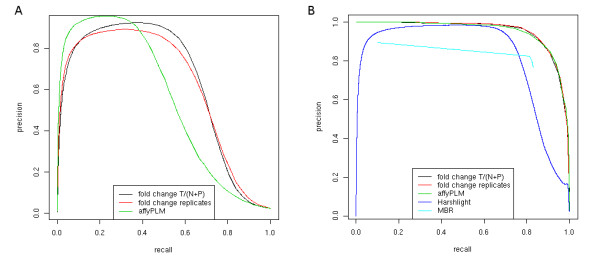
Results: We show that state-of-the-art robust summarization procedures are vulnerable to artefacts on arrays and cannot appropriately correct for these. To address this problem, we present a simple approach to detect artefacts with high recall and precision, which we further improve by taking into account the spatial layout of arrays. Finally, we propose two correction methods for these artefacts that either substitute values of defective probes using probeset information or filter corrupted probes. We show that our approach can identify and correct defective probe measurements appropriately and outperforms existing tools.
Conclusions: While summarization is insufficient to correct for defective probes, this problem can be addressed in a straightforward way by the methods we present for identification and correction of defective probes. As these methods output CEL files with corrected probe values that serve as input to standard normalization and summarization procedures, they can be easily integrated into existing microarray analysis pipelines as an additional pre-processing step. An R package is freely available from http://www.bio.ifi.lmu.de/artefact-correction.
Figures
References
-
- Gardina PJ, Clark TA, Shimada B, Staples MK, Yang Q, Veitch J, Schweitzer A, Awad T, Sugnet C, Dee S, Davies C, Williams A, Turpaz Y. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics. 2006;7:325. doi: 10.1186/1471-2164-7-325. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
