

PyMS: a Python toolkit for processing of gas chromatography-mass spectrometry (GC-MS) data. Application and comparative study of selected tools
- PMID: 22647087
- PMCID: PMC3533878
- DOI: 10.1186/1471-2105-13-115
PyMS: a Python toolkit for processing of gas chromatography-mass spectrometry (GC-MS) data. Application and comparative study of selected tools
Abstract
Background: Gas chromatography-mass spectrometry (GC-MS) is a technique frequently used in targeted and non-targeted measurements of metabolites. Most existing software tools for processing of raw instrument GC-MS data tightly integrate data processing methods with graphical user interface facilitating interactive data processing. While interactive processing remains critically important in GC-MS applications, high-throughput studies increasingly dictate the need for command line tools, suitable for scripting of high-throughput, customized processing pipelines.
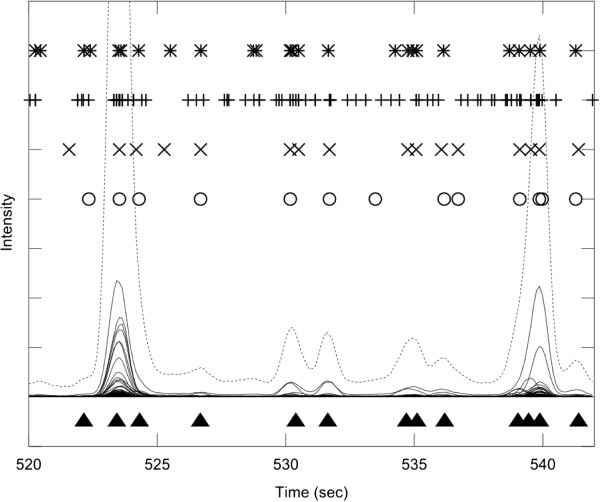
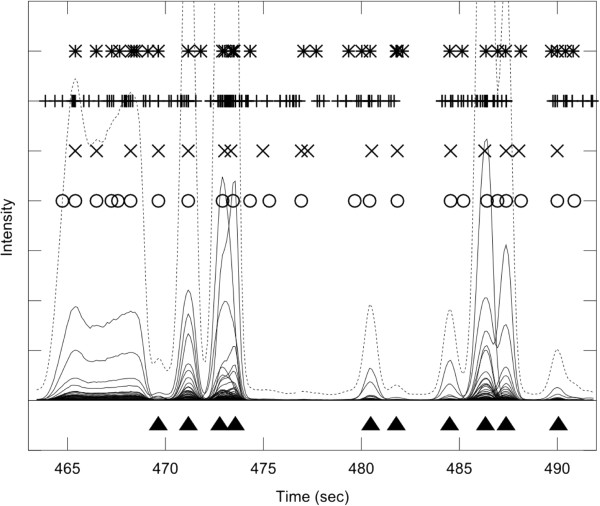
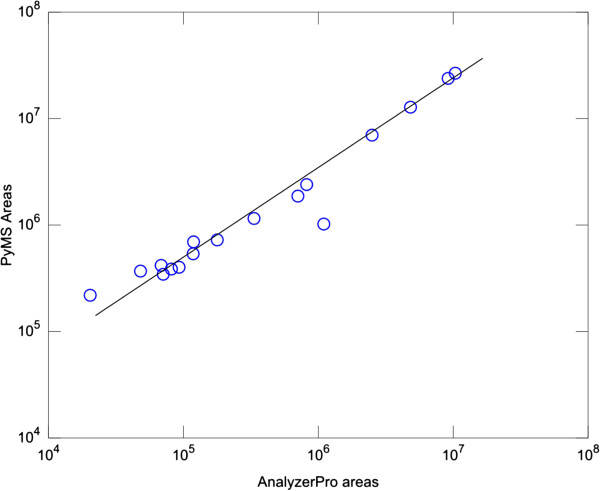
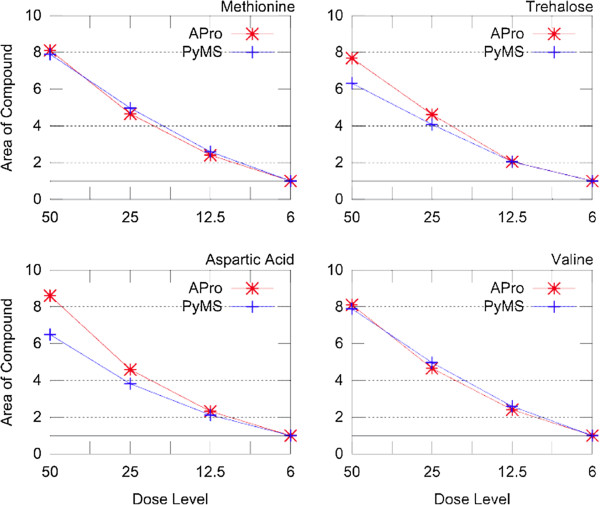
Results: PyMS comprises a library of functions for processing of instrument GC-MS data developed in Python. PyMS currently provides a complete set of GC-MS processing functions, including reading of standard data formats (ANDI- MS/NetCDF and JCAMP-DX), noise smoothing, baseline correction, peak detection, peak deconvolution, peak integration, and peak alignment by dynamic programming. A novel common ion single quantitation algorithm allows automated, accurate quantitation of GC-MS electron impact (EI) fragmentation spectra when a large number of experiments are being analyzed. PyMS implements parallel processing for by-row and by-column data processing tasks based on Message Passing Interface (MPI), allowing processing to scale on multiple CPUs in distributed computing environments. A set of specifically designed experiments was performed in-house and used to comparatively evaluate the performance of PyMS and three widely used software packages for GC-MS data processing (AMDIS, AnalyzerPro, and XCMS).
Conclusions: PyMS is a novel software package for the processing of raw GC-MS data, particularly suitable for scripting of customized processing pipelines and for data processing in batch mode. PyMS provides limited graphical capabilities and can be used both for routine data processing and interactive/exploratory data analysis. In real-life GC-MS data processing scenarios PyMS performs as well or better than leading software packages. We demonstrate data processing scenarios simple to implement in PyMS, yet difficult to achieve with many conventional GC-MS data processing software. Automated sample processing and quantitation with PyMS can provide substantial time savings compared to more traditional interactive software systems that tightly integrate data processing with the graphical user interface.
Figures
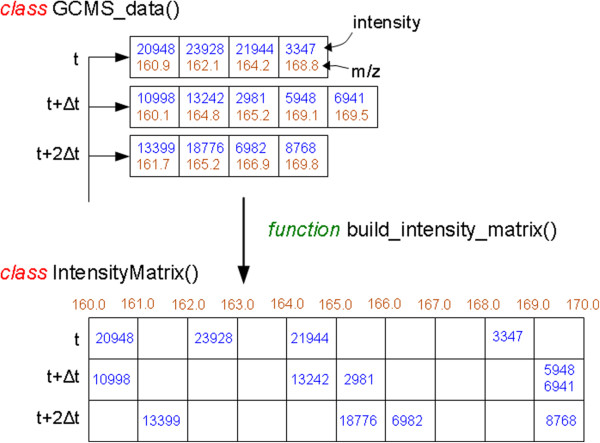
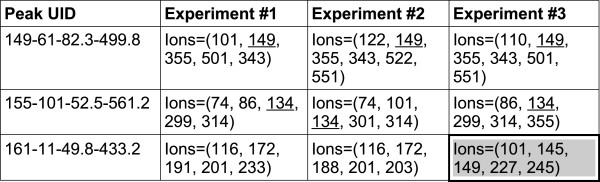
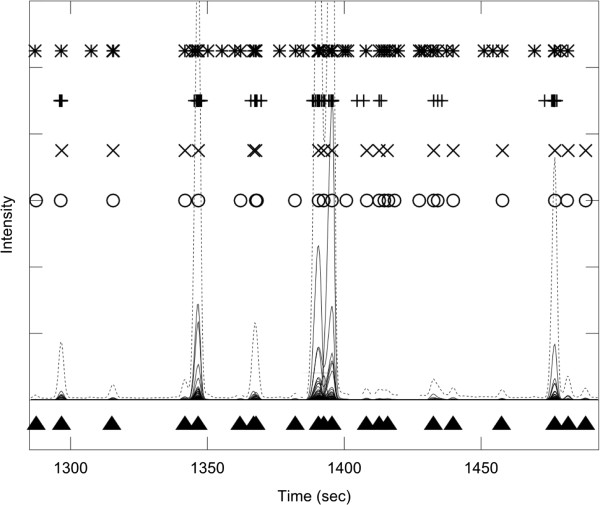
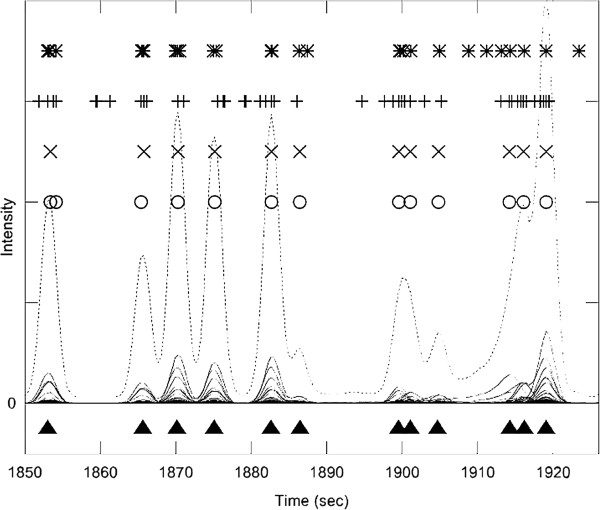
References
-
- Halket JM, Waterman D, Przyborowska AM, Patel RK, Fraser PD, Bramley PM. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J Exp Bot. 2005;56(410):219–243. - PubMed
-
- Kopka J. In: Plant Metabolomics. Saito K, Dixon RA, Willmitzer L, editor. Berlin Heidelberg: Springer; 2006. Gas Chromatography Mass Spectrometry; pp. 3–20.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
